Mitochondrial BK Channel Openers CGS7181 and CGS7184 Exhibit Cytotoxic Properties

Abstract
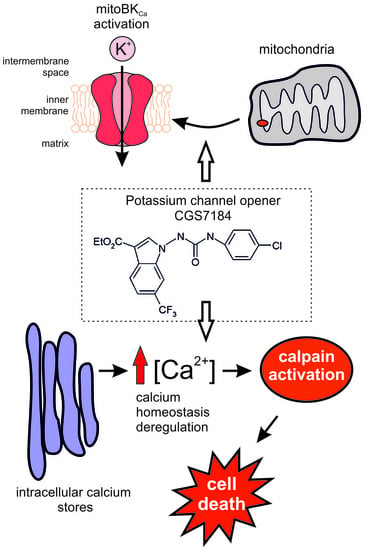
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
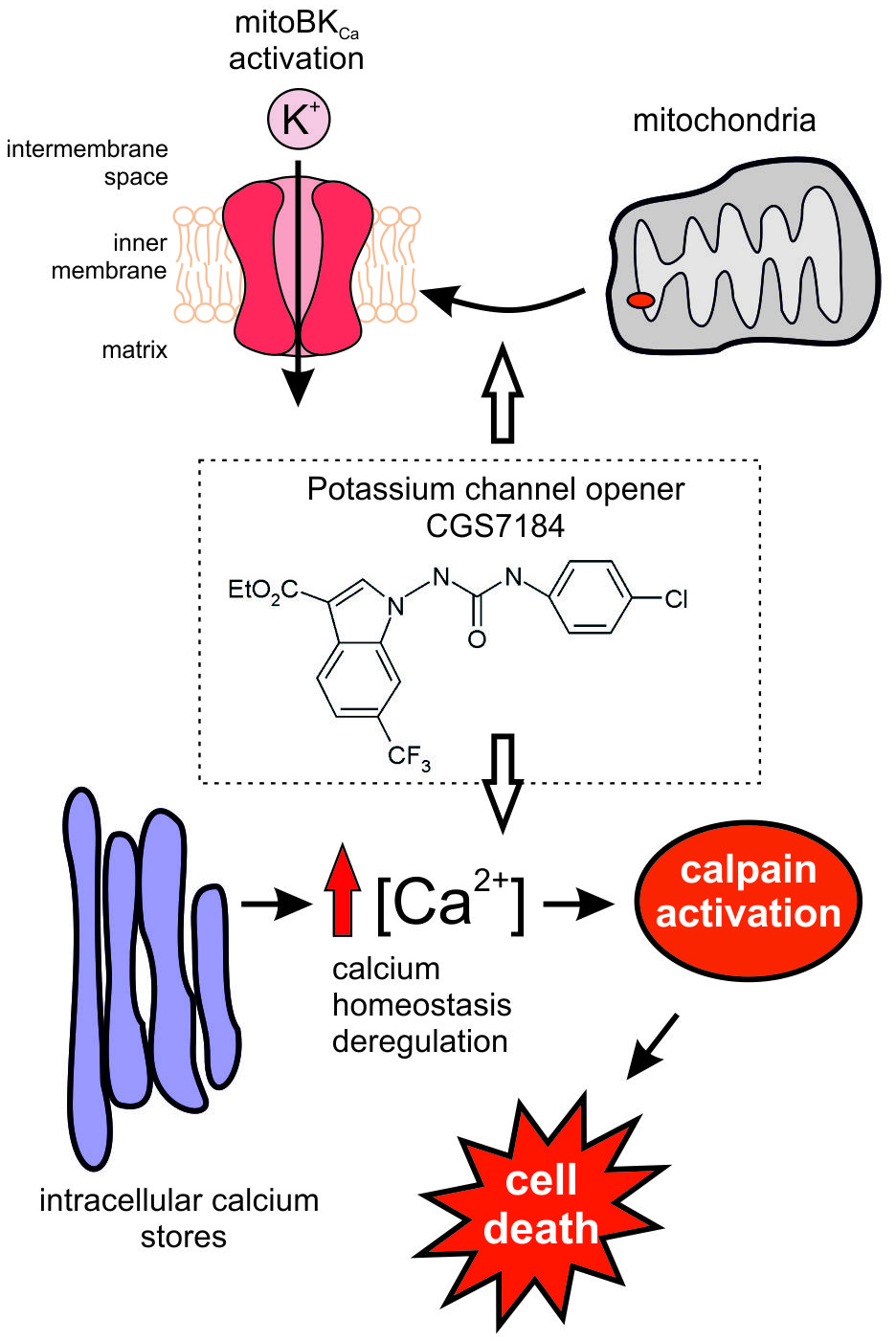
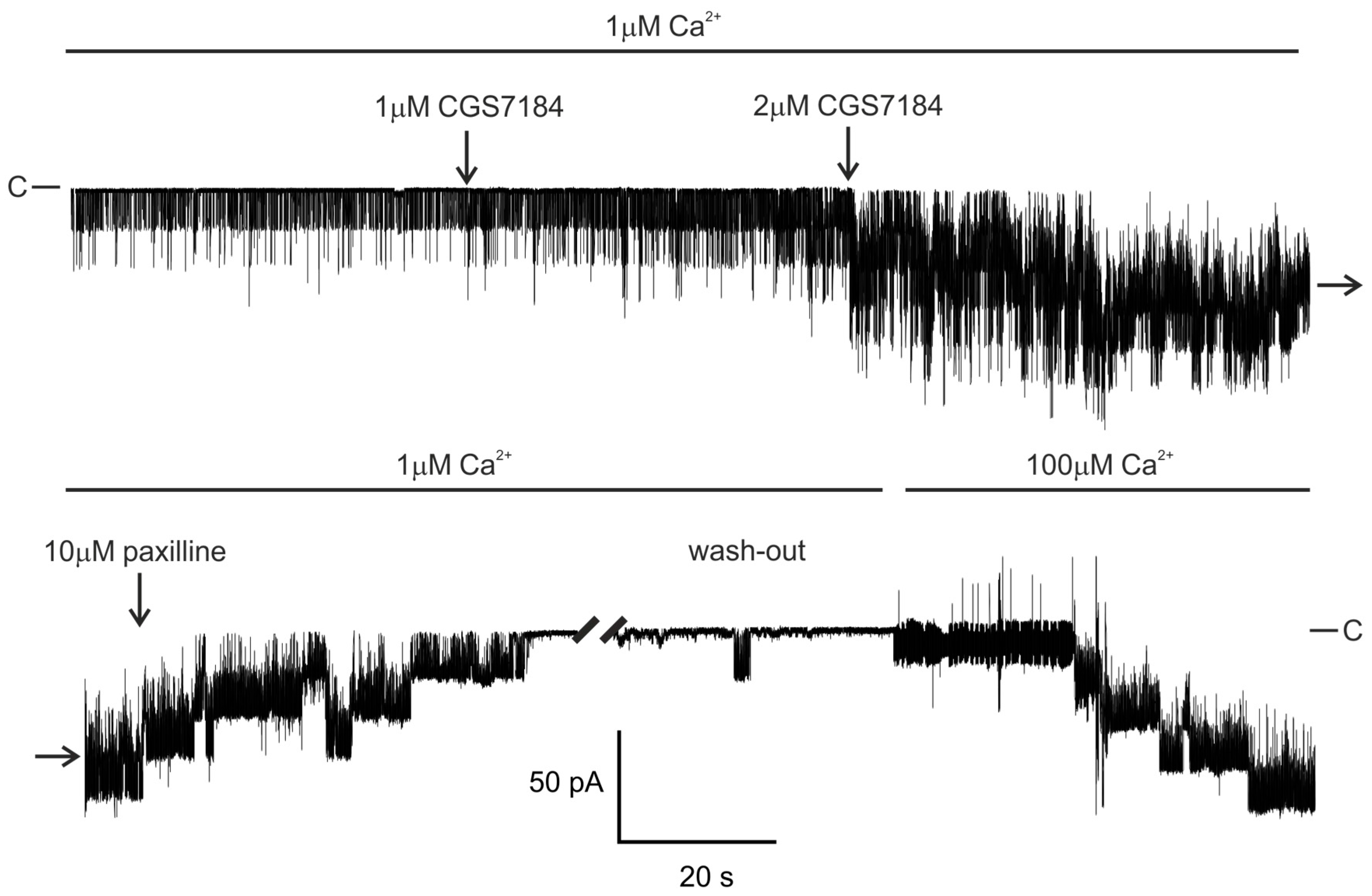
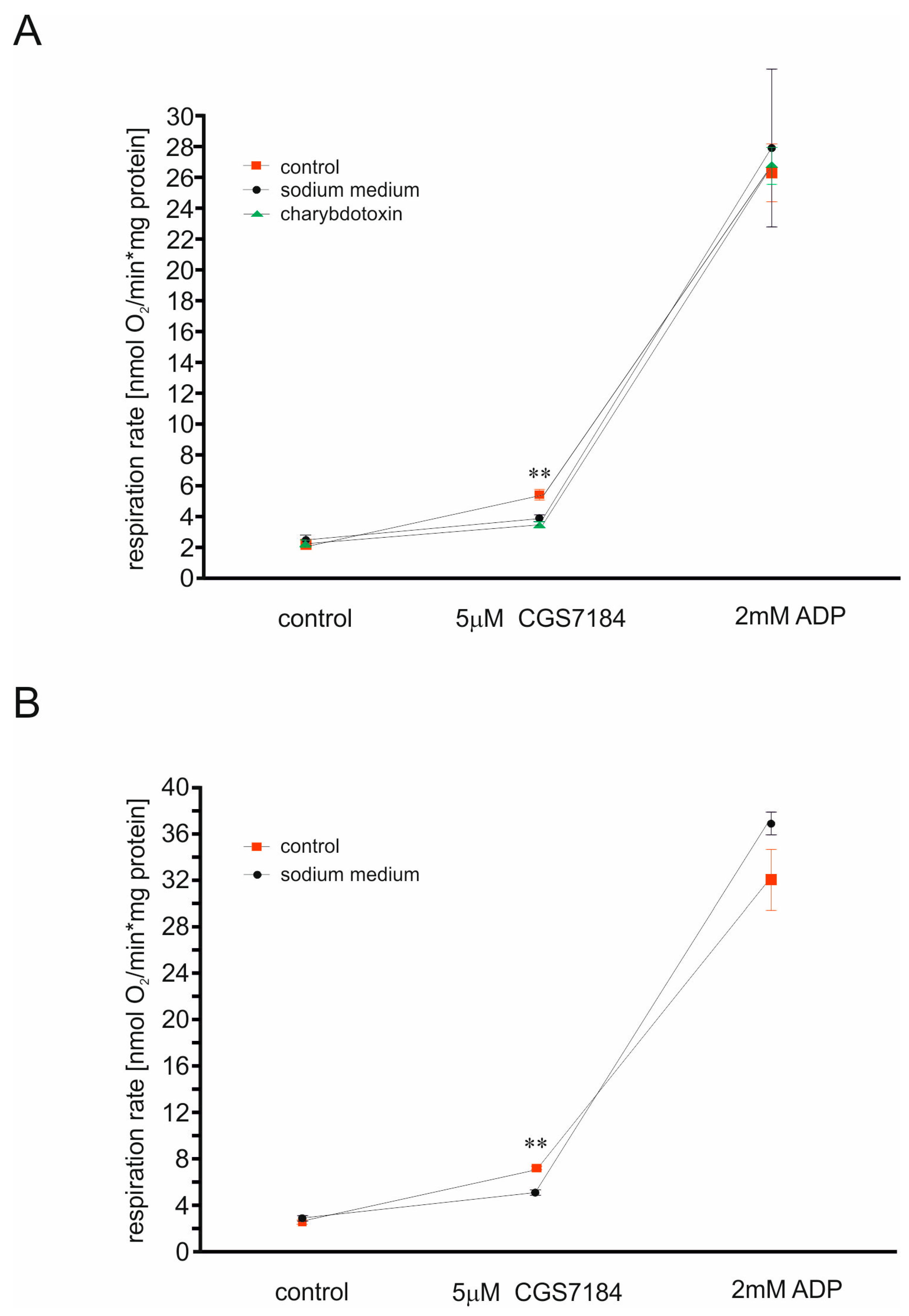
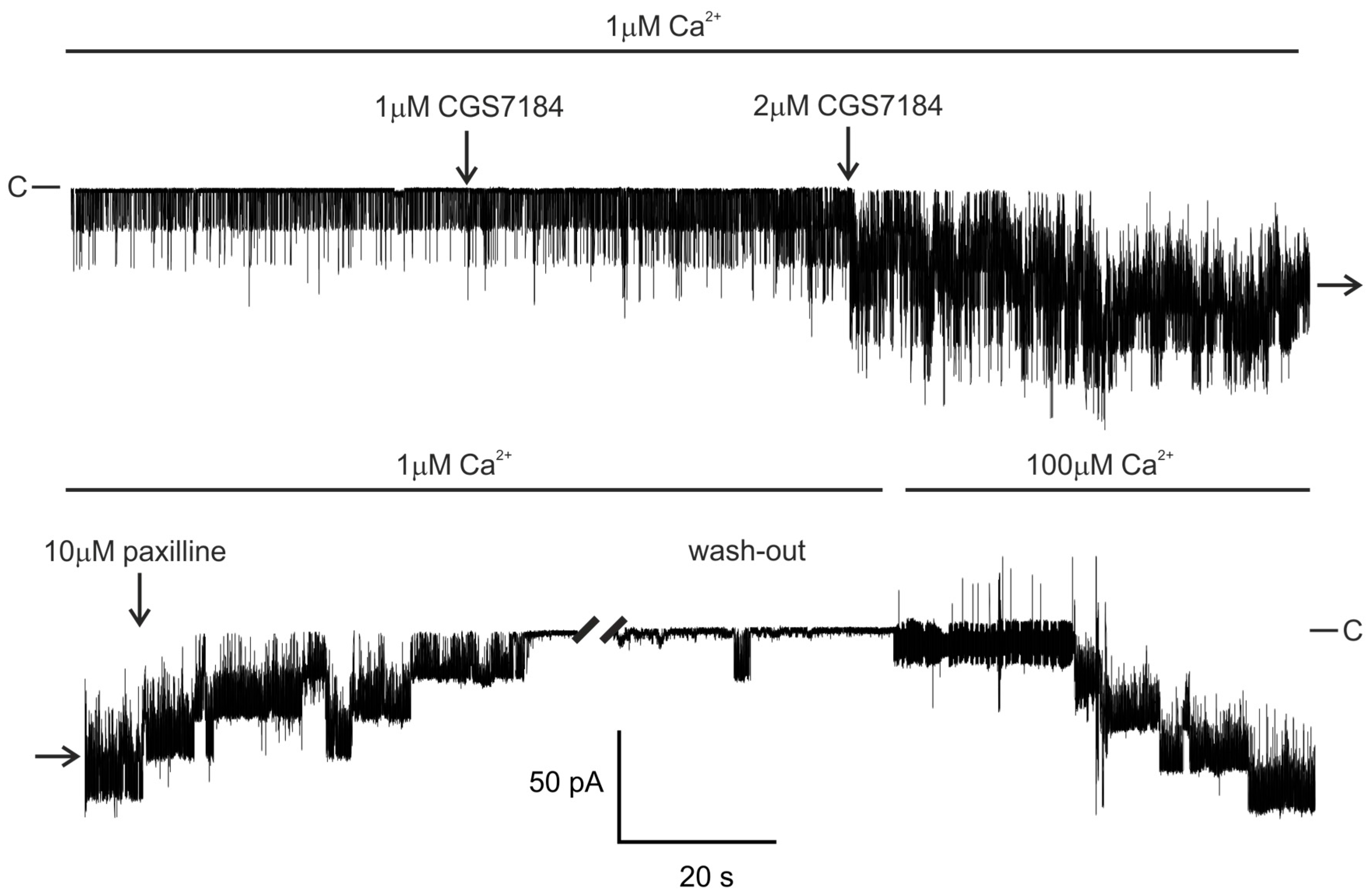
2.1. CGS7184 Activates mitoBKCa
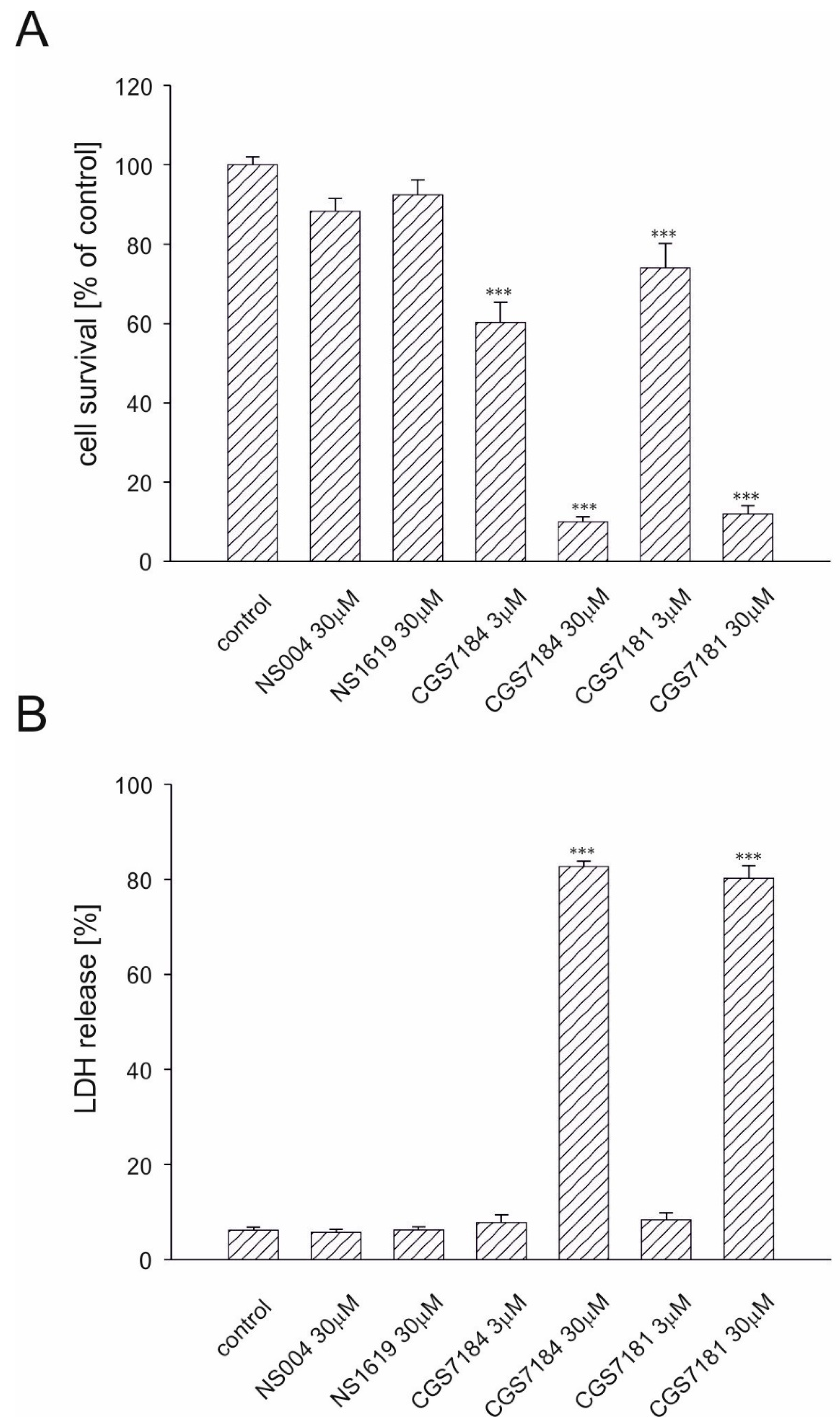
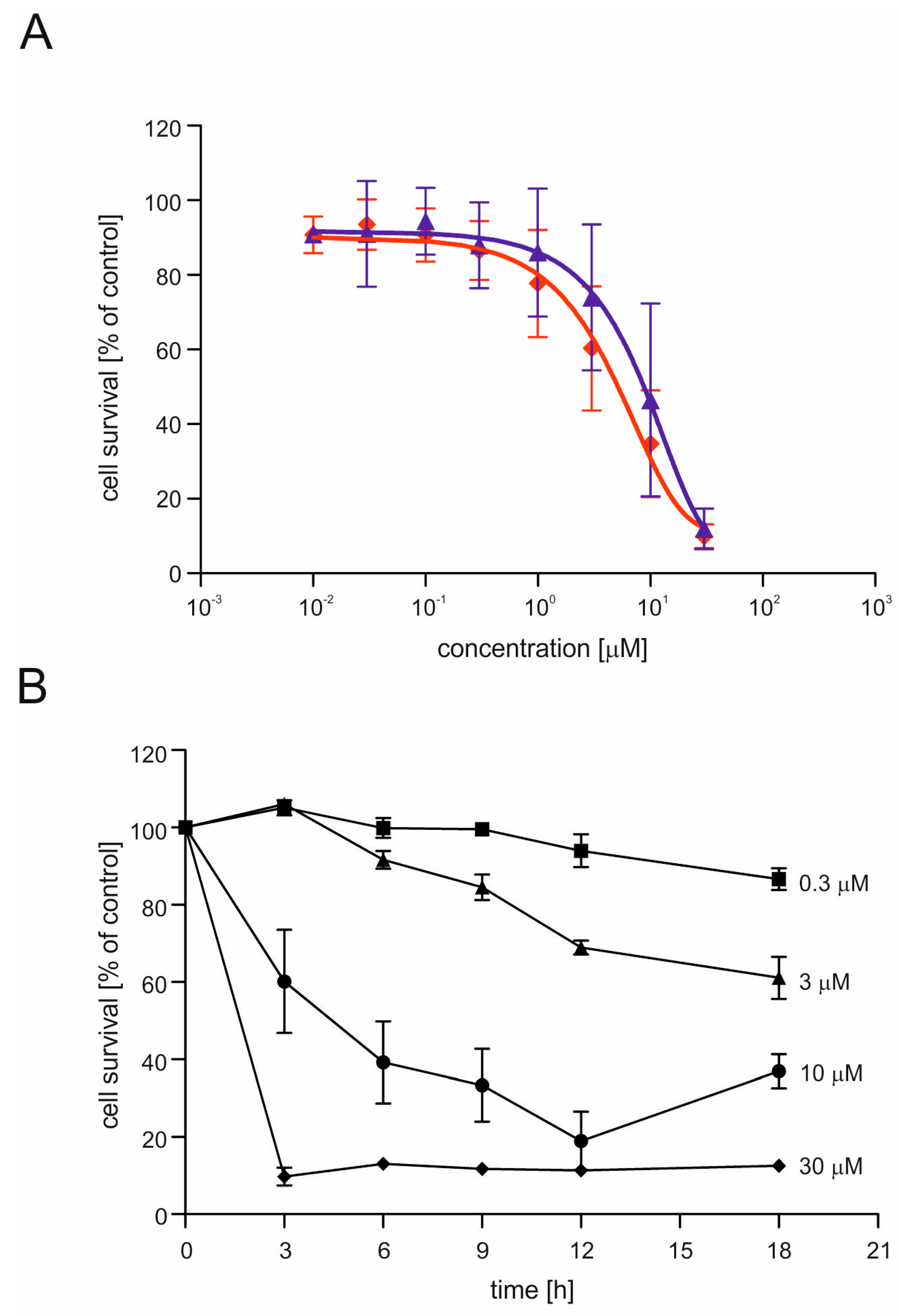
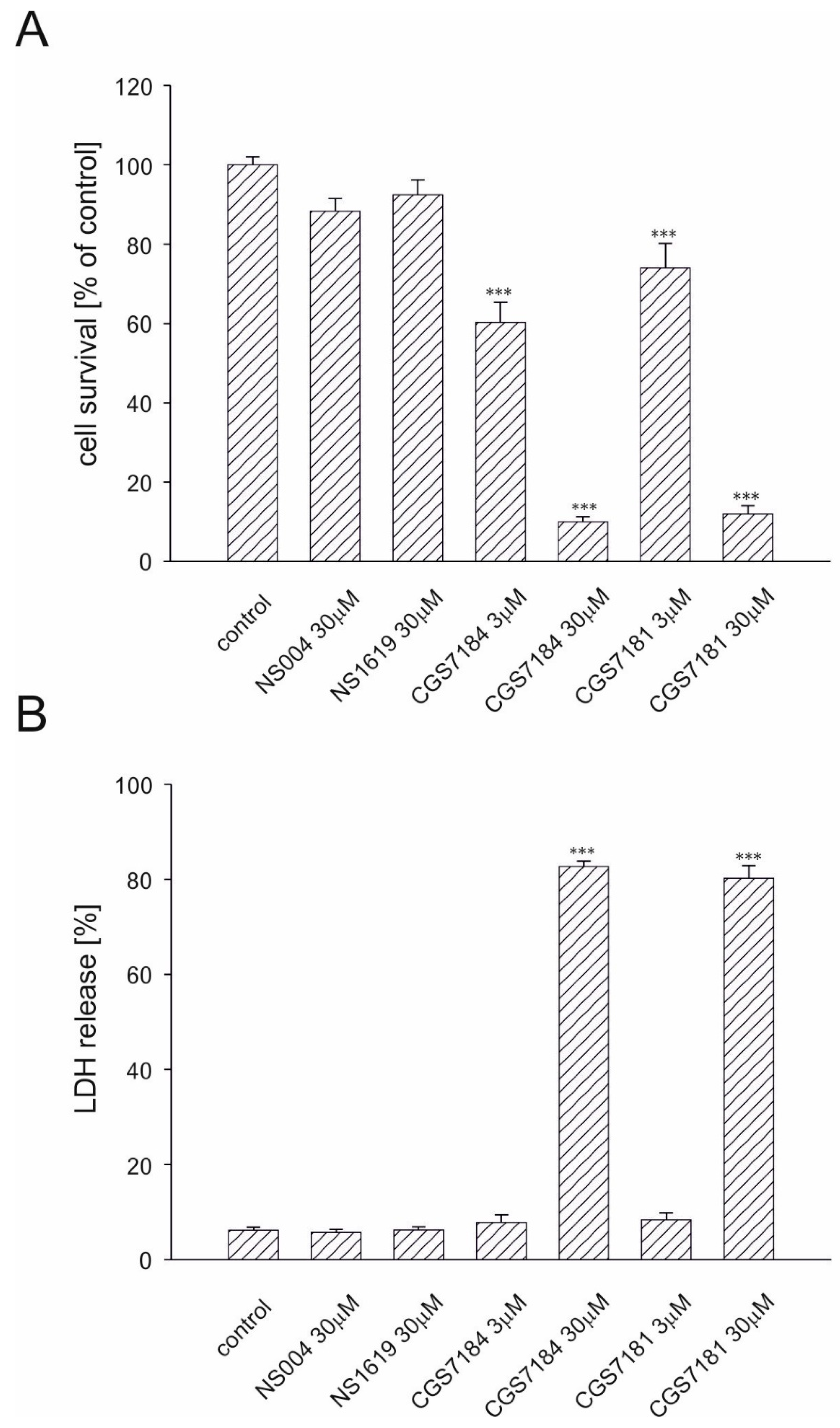
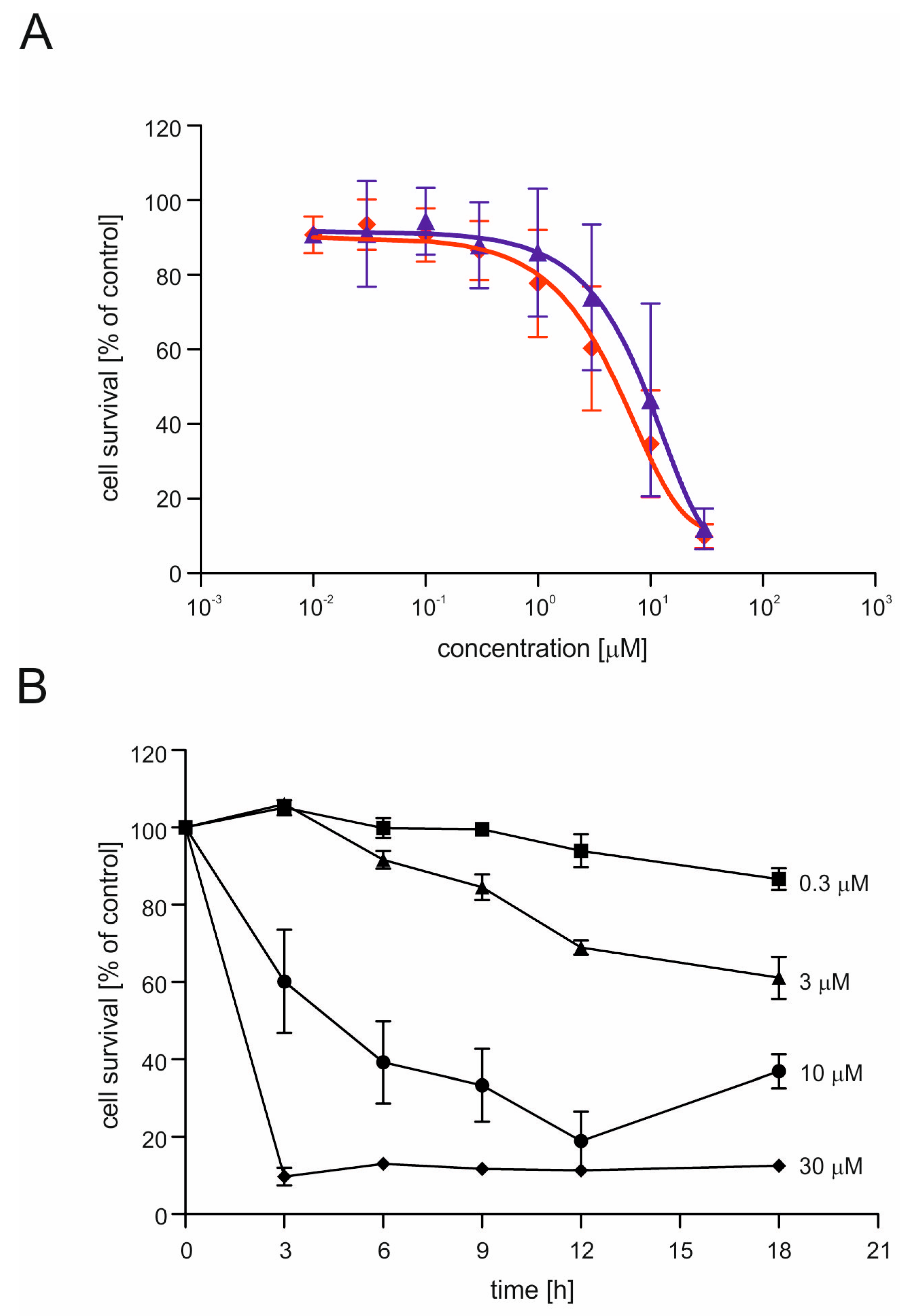
2.2. CGS7181 and CGS7184 Induce HT22 Cell Death in a Dose- and Time-Dependent Manner
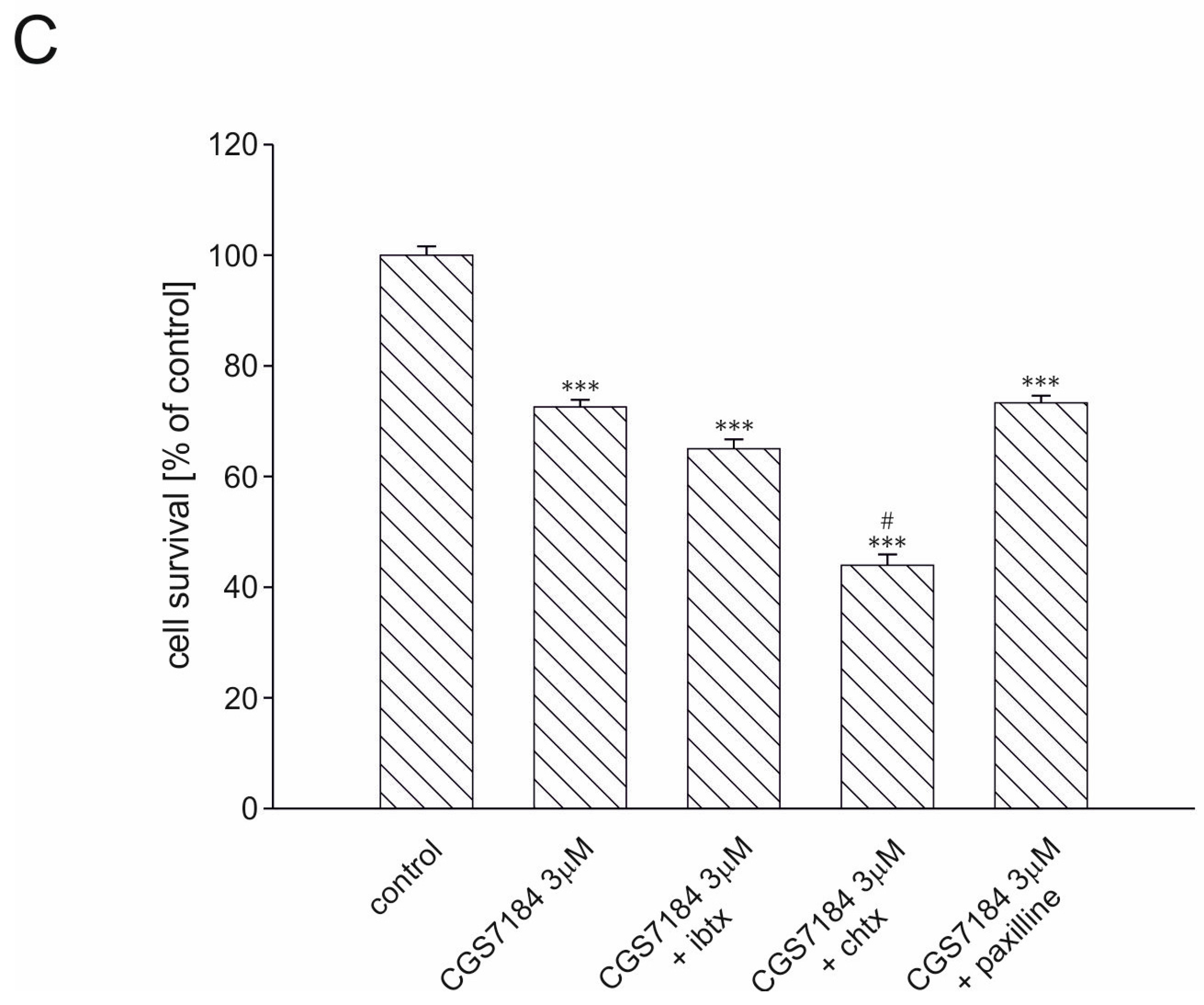
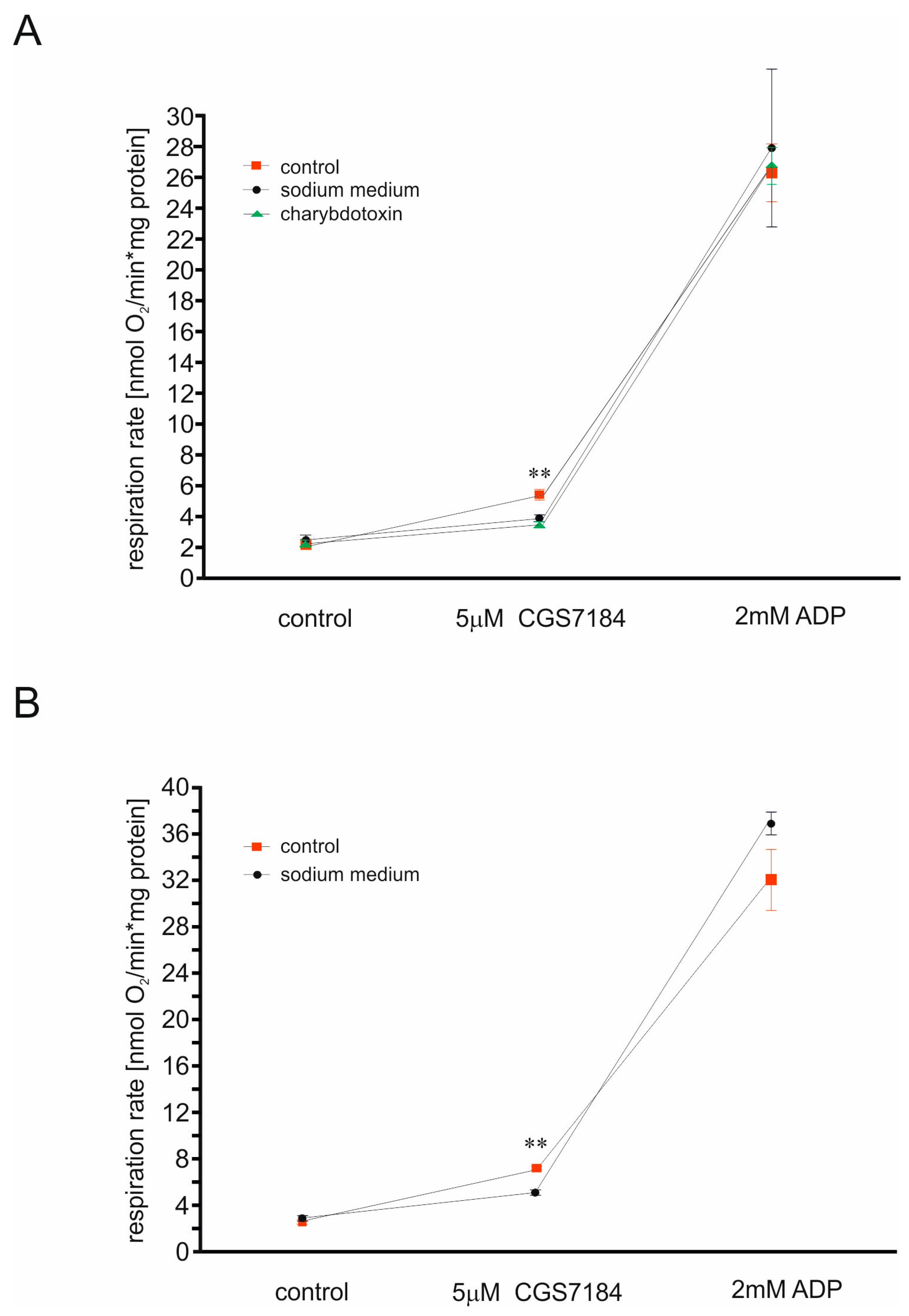
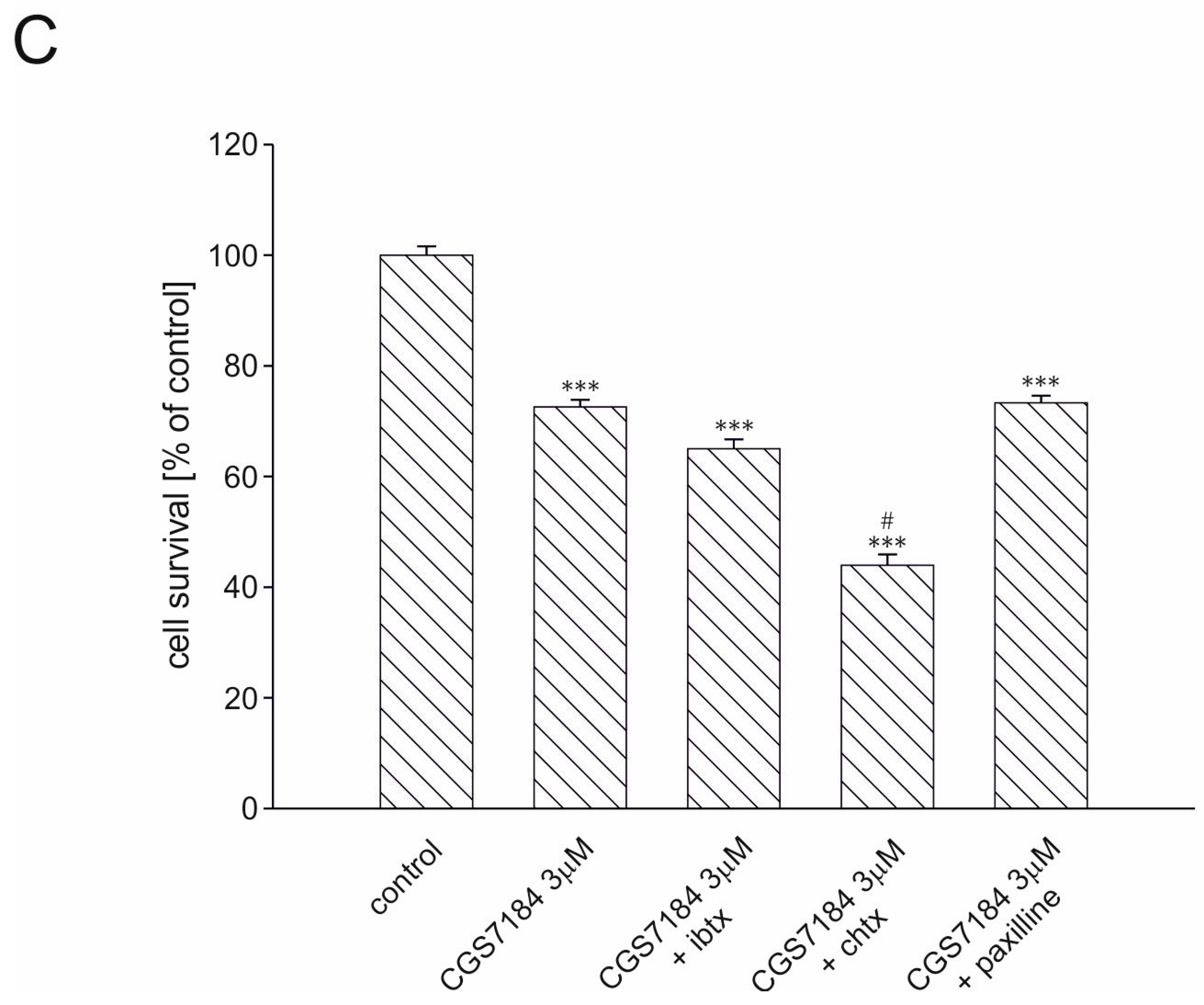
2.3. BKCa Channels Are Not Involved in CGS7184-Induced HT22 Cell Death
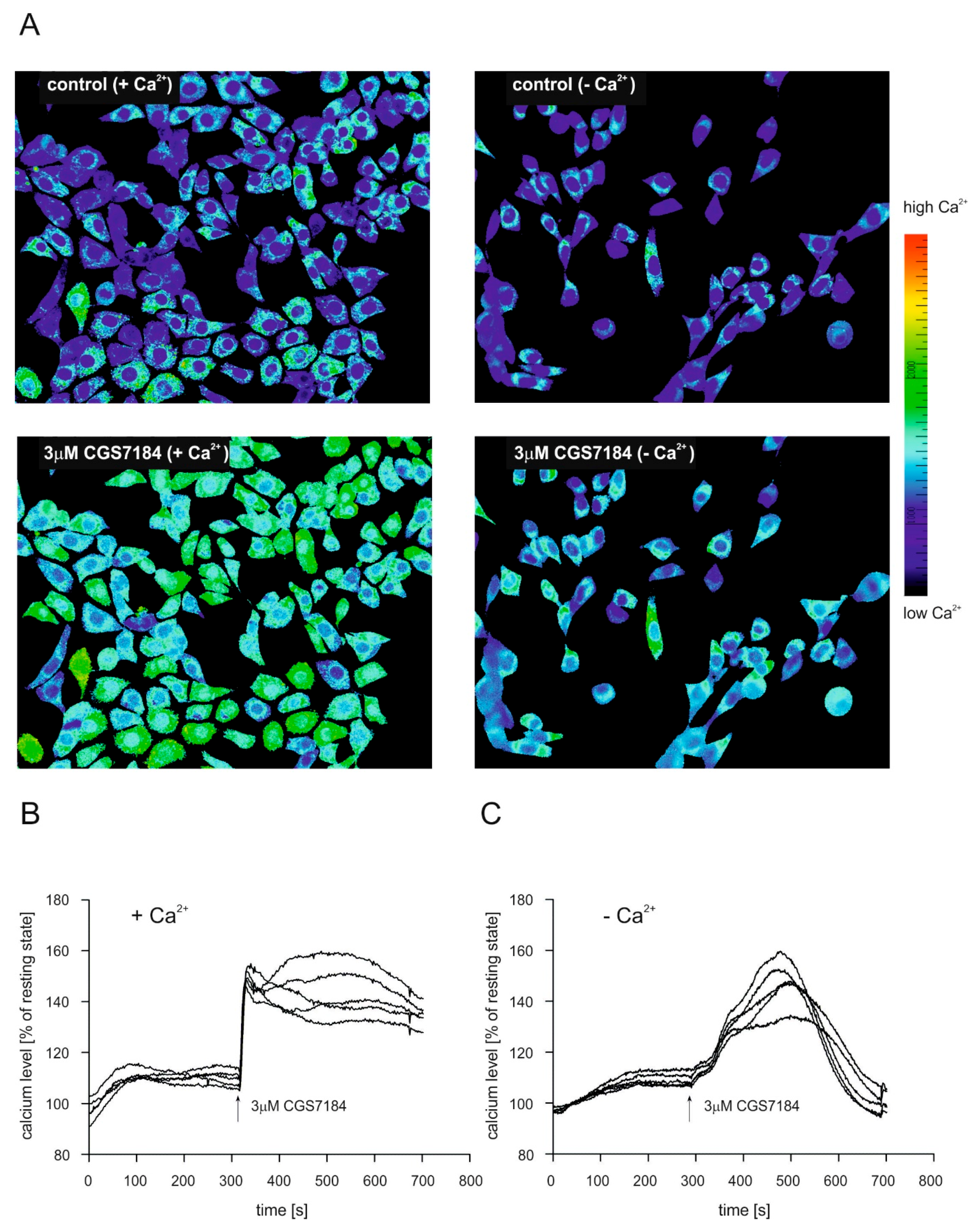
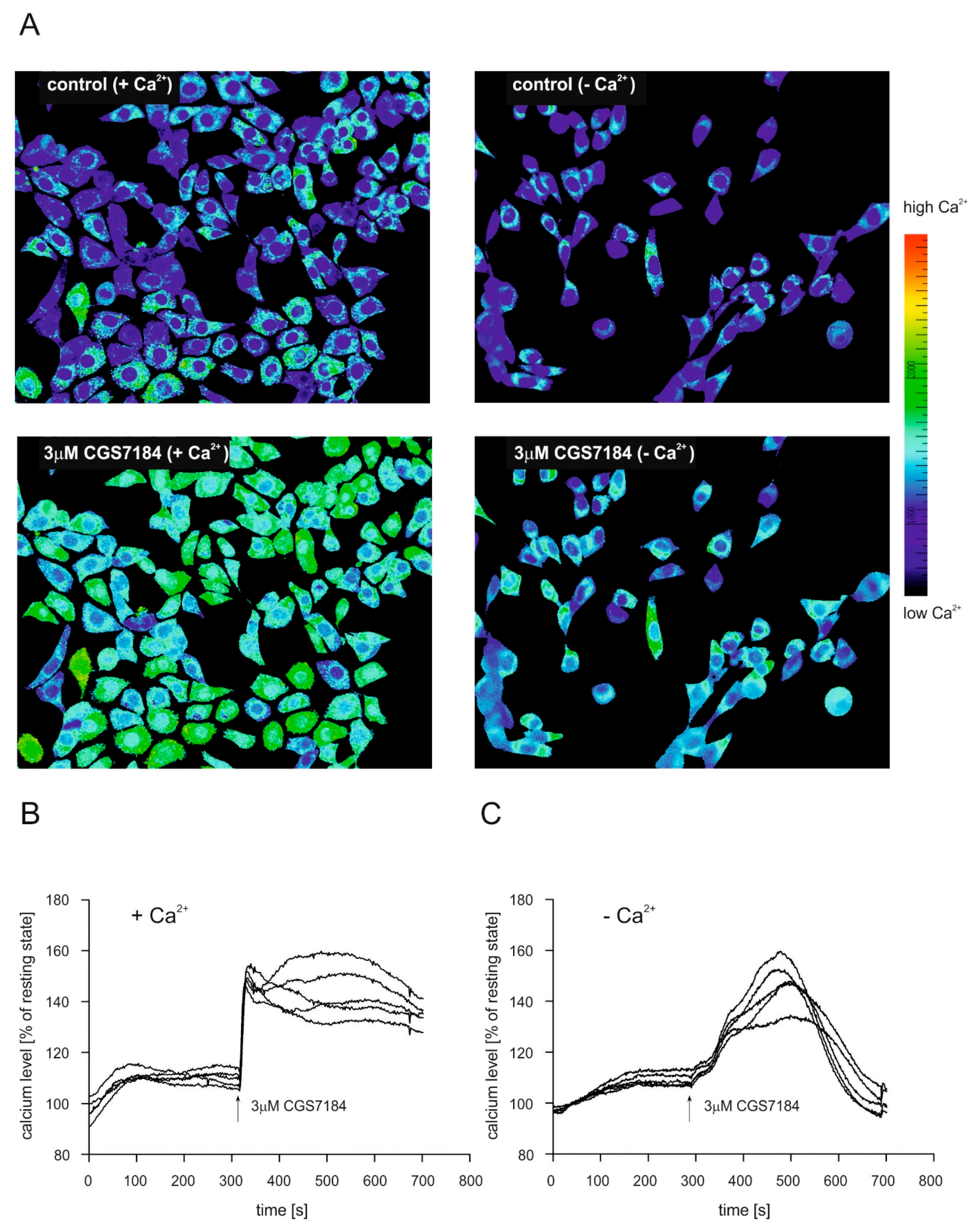
2.4. CGS7184 Elevates Cytoplasmic Calcium Concentration
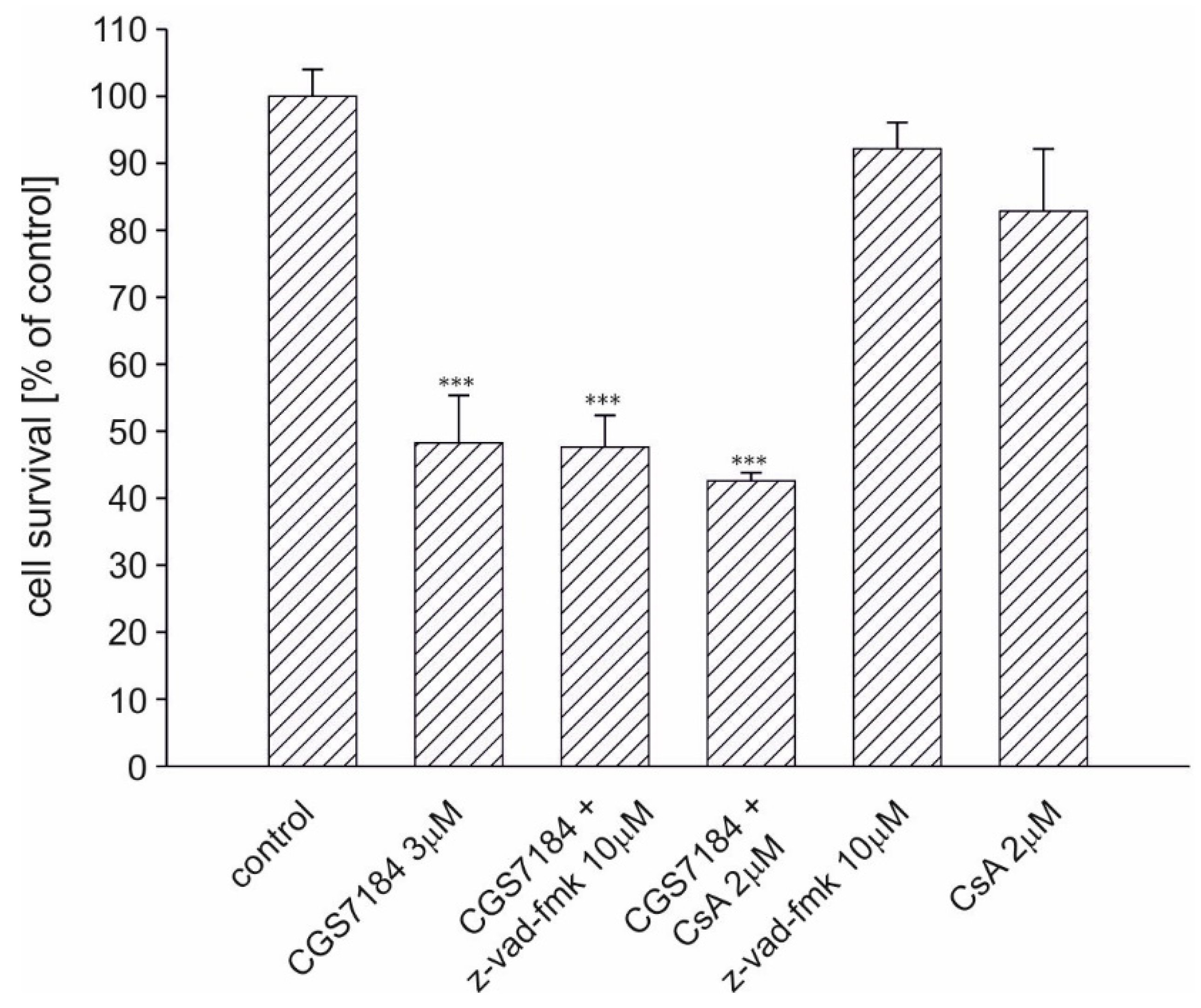
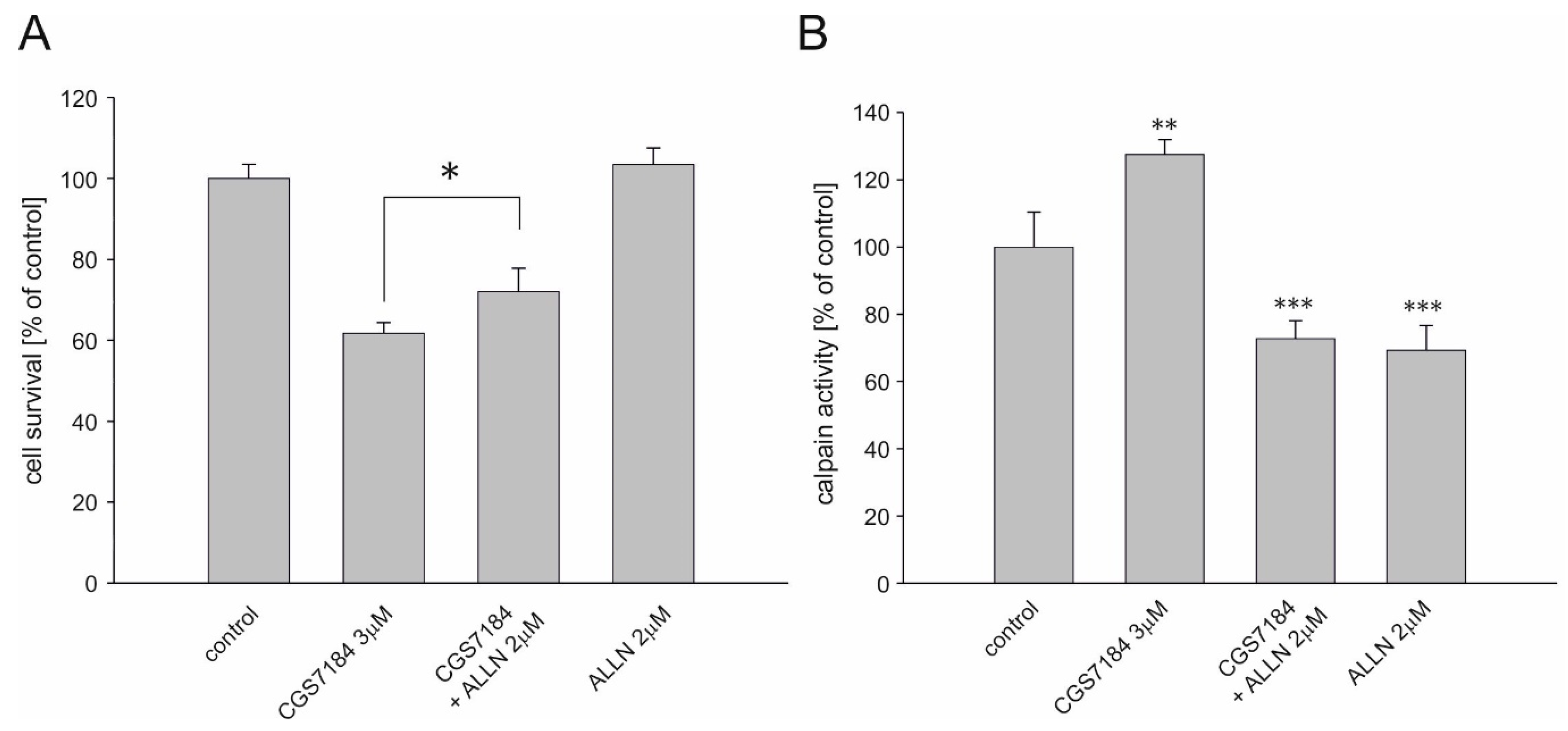
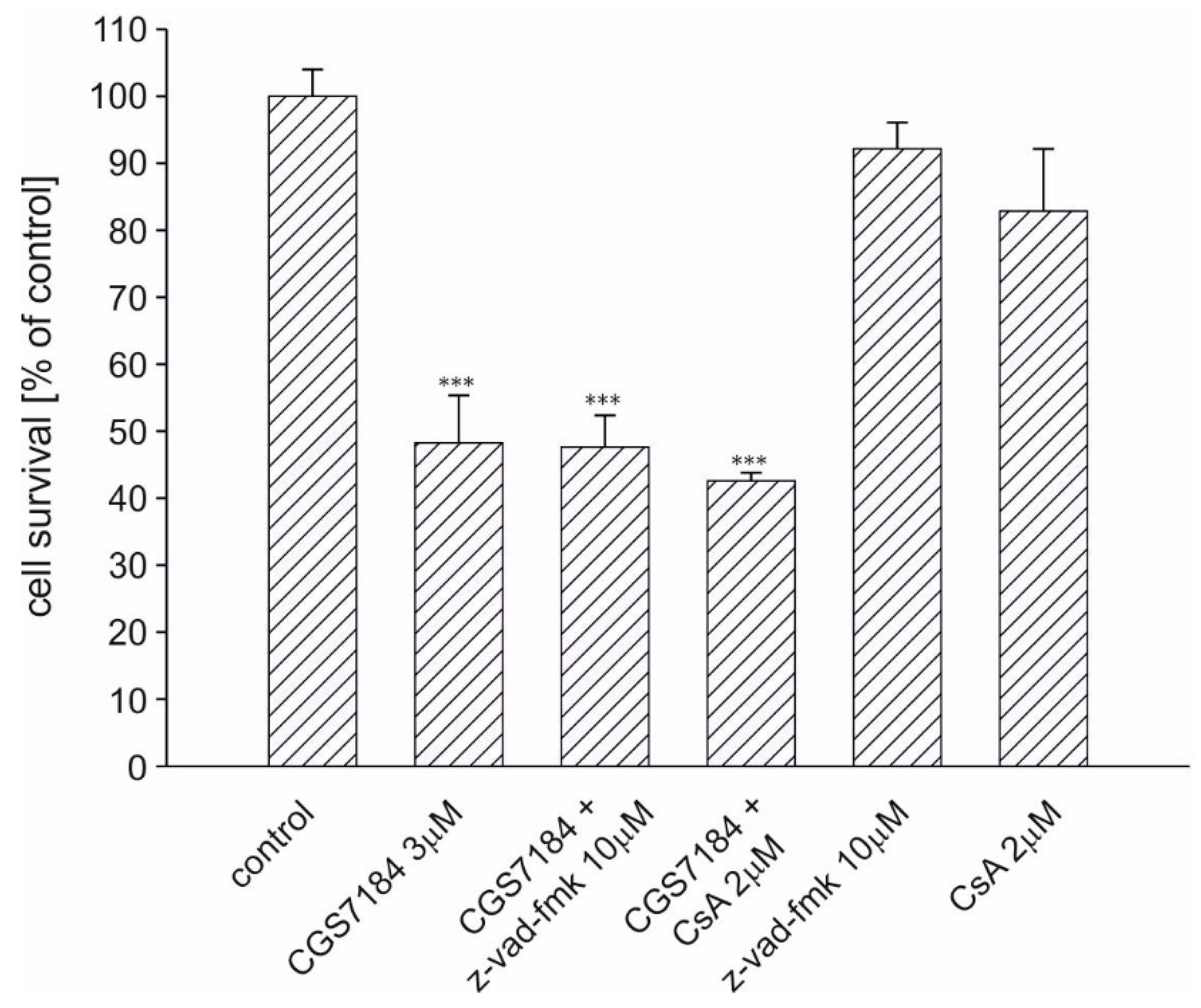
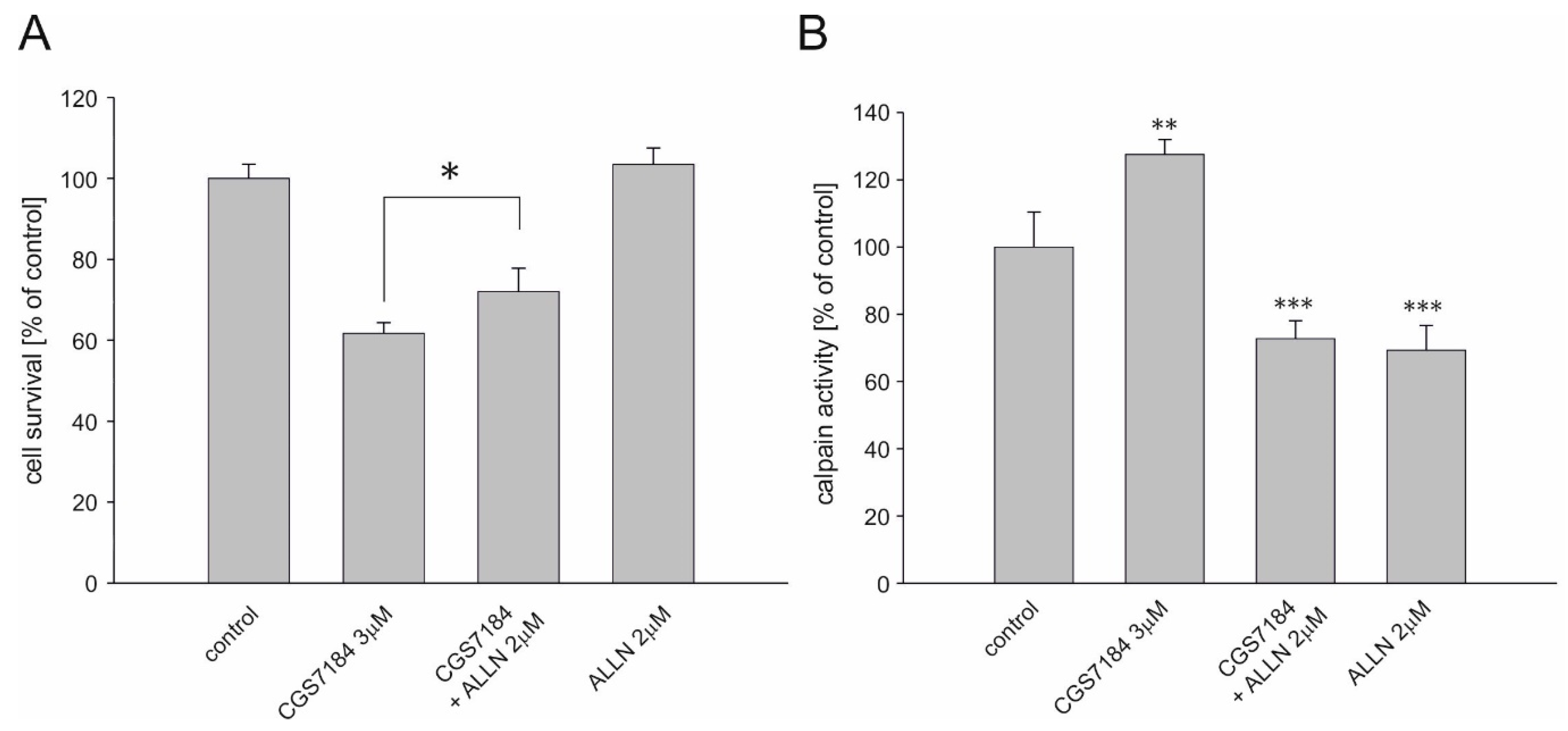
2.5. Calpain Activation Mediates CGS7184 Toxicity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Electrophysiology
4.3. Rat Brain Mitochondria Isolation
4.4. Rat Brain Homogenate Preparation
4.5. Oxygen Consumption Rate
4.6. Cell Culture
4.7. MTT Cell Viability Assay and Lactate Dehydrogenase Assay
4.8. Fluorescence Measurements of Ca2+ Concentration in Single Cells
4.9. Calpain Activity Measurements
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IbTx | Iberiotoxin |
| Pax | Paxilline |
| KCOs | Potassium channel openers |
| PTP | Permeability transition pore |
References
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Knaus, H.G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garciacalvo, M.; Helms, L.M.H.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P.; Smith, A.B.; et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.L.; Fink, C.A.; Kim, H.S.; Lappe, R.W. Novel and potent BK channel openers: CGS 7181 and its analogs. Drug Dev. Res. 1997, 41, 10–21. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Nardi, A.; Calloe, K.; Madsen, L.S.; Olesen, S.P.; Grunnet, M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol. Pharmacol. 2007, 72, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Calderone, V.; Spogli, R.; Martelli, A.; Manfroni, G.; Testai, L.; Sabatini, S.; Tabarrini, O.; Cecchetti, V. Novel 1,4-benzothiazine derivatives as large conductance Ca2+-activated potassium channel openers. J. Med. Chem. 2008, 51, 5085–5092. [Google Scholar] [CrossRef] [PubMed]
- Nardi, A.; Olesen, S.P. BK channel modulators: A comprehensive overview. Curr. Med. Chem. 2008, 15, 1126–1146. [Google Scholar] [CrossRef] [PubMed]
- Facundo, H.T.F.; Fornazari, M.; Kowaltowski, A.J. Tissue protection mediated by mitochondrial K+ channels. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.H.; Liu, Y.G.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Saito, T.; Saegusa, N.; Nakaya, H. Mitochondrial Ca2+-activated K+ channels in cardiac myocytes—A mechanism of the cardioprotective effect and modulation by protein kinase A. Circulation 2005, 111, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Siemen, D.; Loupatatzis, C.; Borecky, J.; Gulbins, E.; Lang, F. Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 1999, 257, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Skalska, J.; Piwonska, M.; Wyroba, E.; Surmacz, L.; Wieczorek, R.; Koszela-Piotrowska, I.; Zielinska, J.; Bednarczyk, P.; Dolowy, K.; Wilczynski, G.M.; et al. A novel potassium channel in skeletal muscle mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Douglas, R.M.; Lai, J.C.K.; Bian, S.; Cummins, L.; Moczydlowski, E.; Haddad, G.G. The calcium-sensitive large-conductance potassium channel (BK/Maxi K) is present in the inner mitochondrial membrane of rat brain. Neuroscience 2006, 139, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Piwonska, M.; Wilczek, E.; Szewczyk, A.; Wilczynski, G.M. Differential distribution of Ca2+-activated potassium channel beta 4 subunit in rat brain: Immunolocalization in neuronal mitochondria. Neuroscience 2008, 153, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Skalska, J.; Bednarczyk, P.; Piwonska, M.; Kulawiak, B.; Wilczynski, G.; Dolowy, K.; Kudin, A.P.; Kunz, W.S.; Szewczyk, A. Calcium ions regulate K+ uptake into brain mitochondria: The evidence for a novel potassium channel. Int. J. Mol. Sci. 2009, 10, 1104–1120. [Google Scholar] [CrossRef] [PubMed]
- Heinen, A.; Aldakkak, M.; Stowe, D.F.; Rhodes, S.S.; Riess, M.L.; Varadarajan, S.G.; Camara, A.K.S. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1400–H1407. [Google Scholar] [CrossRef] [PubMed]
- Kulawiak, B.; Kudin, A.P.; Szewczyk, A.; Kunz, W.S. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp. Neurol. 2008, 212, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, A.; Kajma, A.; Malinska, D.; Wrzosek, A.; Bednarczyk, P.; Zablocka, B.; Dolowy, K. Pharmacology of mitochondrial potassium channels: Dark side of the field. FEBS Lett. 2010, 584, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.; Katakam, P.; Snipes, J.A.; Kis, B.; Domoki, F.; Bari, F.; Busija, D.W. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J. Neurochem. 2008, 105, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.; Domoki, F.; Lenti, L.; Katakam, P.V.G.; Snipes, J.A.; Bari, F.; Busija, D.W. Immediate neuronal preconditioning by NS1619. Brain Res. 2009, 1285, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Debska, G.; Kicinska, A.; Dobrucki, J.; Dworakowska, B.; Nurowska, E.; Skalska, J.; Dolowy, K.; Szewczyk, A. Large-conductance K+ channel openers NS1619 and NS004 as inhibitors of mitochondrial function in glioma cells. Biochem. Pharmacol. 2003, 65, 1827–1834. [Google Scholar] [CrossRef]
- Wrzosek, A.; Lukasiak, A.; Gwozdz, P.; Malinska, D.; Kozlovski, V.I.; Szewczyk, A.; Chlopicki, S.; Dolowy, K. Large-conductance K+ channel opener CGS7184 as a regulator of endothelial cell function. Eur. J. Pharmacol. 2009, 602, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, A.; Tomaskova, Z.; Ondrias, K.; Lukasiak, A.; Szewczyk, A. The potassium channel opener CGS7184 activates Ca2+ release from the endoplasmic reticulum. Eur. J. Pharmacol. 2012, 690, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Debska-Vielhaber, G.; Godlewski, M.M.; Kicinska, A.; Skalska, J.; Kulawiak, B.; Piwonska, M.; Zablocki, K.; Kunz, W.S.; Szewczyk, A.; Motyl, T. Large-conductance K+ channel openers induce death of human glioma cells. J. Physiol. Pharmacol. 2009, 60, 27–36. [Google Scholar] [PubMed]
- Bednarczyk, P.; Wieckowski, M.R.; Broszkiewicz, M.; Skowronek, K.; Siemen, D.; Szewczyk, A. Putative structural and functional coupling of the mitochondrial BKCa channel to the respiratory chain. PLoS ONE 2013, 8, e68125. [Google Scholar] [CrossRef] [PubMed]
- Augustynek, B.; Kudin, A.P.; Bednarczyk, P.; Szewczyk, A.; Kunz, W.S. Hemin inhibits the large conductance potassium channel in brain mitochondria: A putative novel mechanism of neurodegeneration. Exp. Neurol. 2014, 257, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Barker, G.D.; Halestrap, A.P. Determination of the rate of K+ movement through potassium channels in isolated rat heart and liver mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Kulawiak, B.; Nencki Institute of Experimental Biology, Warsaw, Poland. Personal communication, 2009.
- Burg, E.D.; Remillard, C.V.; Yuan, J.X.J. K+ stop channels in apoptosis. J. Membr. Biol. 2006, 209, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P.; Yeh, C.H.; Sensi, S.L.; Gwag, B.J.; Canzoniero, L.M.T.; Farhangrazi, Z.S.; Ying, H.S.; Tian, M.; Dugan, L.L.; Choi, D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997, 278, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.M.; Sun, L.N.; Zhou, H.Y.; Wang, X.L. Voltage-dependent potassium channels are involved in glutamate-induced apoptosis of rat hippocampal neurons. Neurosci. Lett. 2006, 398, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.L.; Liu, Z.; Zeng, X.M.; Liu, Z.Q.; Chen, X.H.; Zhang, Z.H.; Mei, Y.A. 4-Aminopyridine, a Kv channel antagonist, prevents apoptosis of rat cerebellar granule neurons. Neuropharmacology 2006, 51, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Piccirilli, S.; Sebastianelli, L.; Nistico, G.; Bernardi, G.; Mercuri, N.B. The blockade of K+-ATP channels has neuroprotective effects in an in vitro model of brain ischemia. In Neuroinflammation in Neuronal Death and Repair; Elsevier: San Diego, CA, USA, 2007; Volume 82, pp. 383–395. [Google Scholar]
- Kulawiak, B.; Szewczyk, A. Glutamate-induced cell death in HT22 mouse hippocampal cells is attenuated by paxilline, a BK channel inhibitor. Mitochondrion 2012, 12, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lukasiak, A.; Skup, A.; Chlopicki, S.; Lomnicka, M.; Kaczara, P.; Proniewski, B.; Szewczyk, A.; Wrzosek, A. SERCA, complex I of the respiratory chain and ATP-synthase inhibition are involved in pleiotropic effects of NS1619 on endothelial cells. Eur. J. Pharmacol. 2016, 786, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, A.L. Apoptosis and necrosis in health and disease: Role of mitochondria. Int. Rev. Cytol. 2003, 224, 29–55. [Google Scholar] [PubMed]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Rao, K.V.R. The mitochondrial permeability transition in neurologic disease. Neurochem. Int. 2007, 50, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Neumar, R.W.; Xu, Y.A.; Gada, H.; Guttmann, R.P.; Siman, R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J. Biol. Chem. 2003, 278, 14162–14167. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sribnick, E.A.; Wingrave, J.M.; del Re, A.M.; Woodward, J.J.; Appel, S.H.; Banik, N.L.; Ray, S.K. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: Calpain inhibition provides functional neuroprotection. J. Neurosci. Res. 2005, 81, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Marcilhac, A. Implication of calpain in neuronal apoptosis—A possible regulation of Alzheimer’s disease. FEBS J. 2006, 273, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.K.; Karmakar, S.; Nowak, M.W.; Banik, N.L. Inhibition of calpain and caspase-3 prevented apoptosis and preserved electrophysiological properties of voltage-gated and ligand-gated ion channels in rat primary cortical neurons exposed to glutamate. Neuroscience 2006, 139, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, K.; Leist, M.; Groc, L. Pathological apoptosis in the developing brain. Apoptosis 2007, 12, 993–1010. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Schnellmann, R.G. Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 2012, 96, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Walewska, A.; Koprowski, P.; Szewczyk, A. Mechanosensitivity of Mitochondrial Calcium-Activated Potassium Channel of Large Conductance (mitoBKCa). Unpublished work. 2017. [Google Scholar]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augustynek, B.; Koprowski, P.; Rotko, D.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Mitochondrial BK Channel Openers CGS7181 and CGS7184 Exhibit Cytotoxic Properties. Int. J. Mol. Sci. 2018, 19, 353. https://doi.org/10.3390/ijms19020353
Augustynek B, Koprowski P, Rotko D, Kunz WS, Szewczyk A, Kulawiak B. Mitochondrial BK Channel Openers CGS7181 and CGS7184 Exhibit Cytotoxic Properties. International Journal of Molecular Sciences. 2018; 19(2):353. https://doi.org/10.3390/ijms19020353
Chicago/Turabian StyleAugustynek, Bartłomiej, Piotr Koprowski, Daria Rotko, Wolfram S. Kunz, Adam Szewczyk, and Bogusz Kulawiak. 2018. "Mitochondrial BK Channel Openers CGS7181 and CGS7184 Exhibit Cytotoxic Properties" International Journal of Molecular Sciences 19, no. 2: 353. https://doi.org/10.3390/ijms19020353