HDAC Inhibition Improves the Sarcoendoplasmic Reticulum Ca2+-ATPase Activity in Cardiac Myocytes

 , , , , , , ,
, , , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
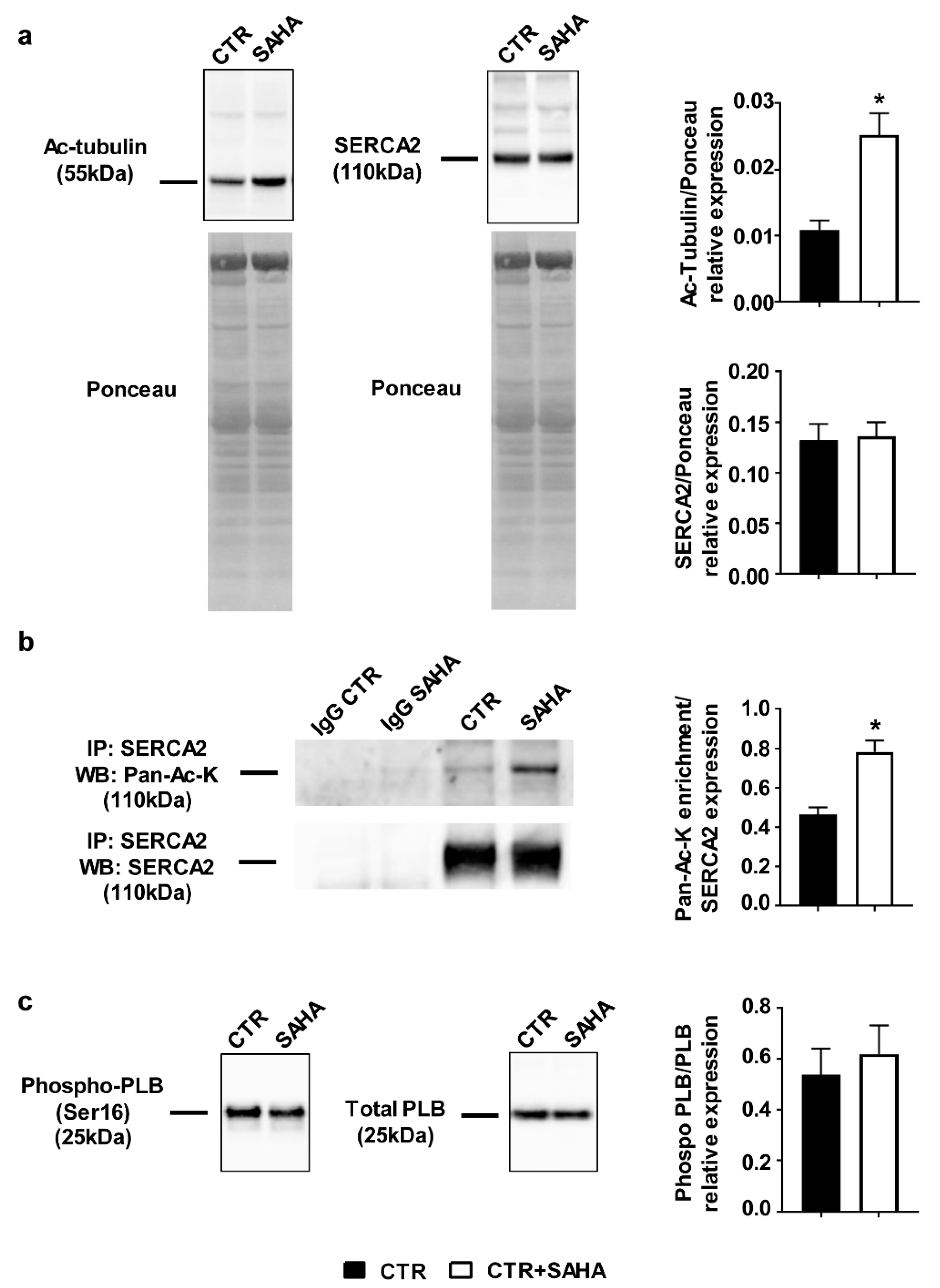
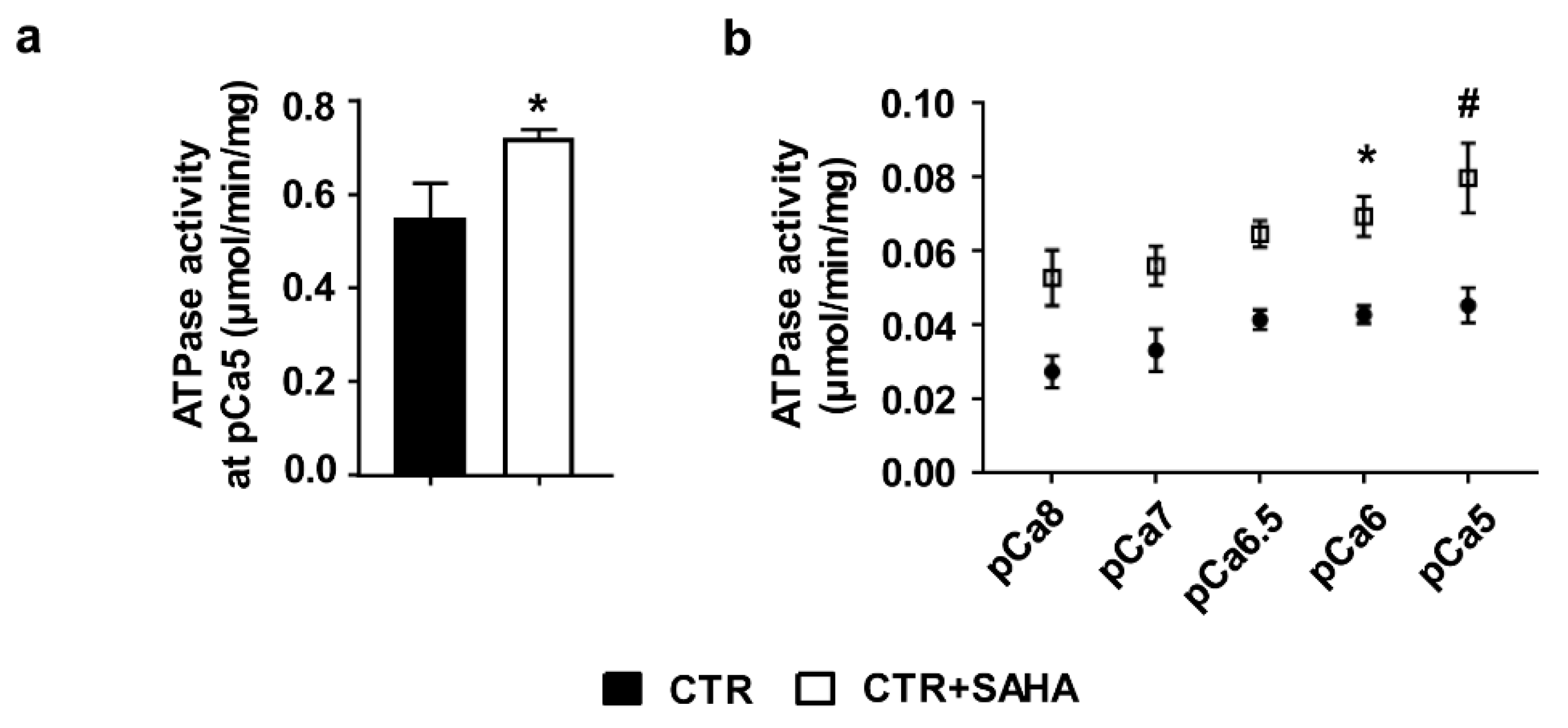
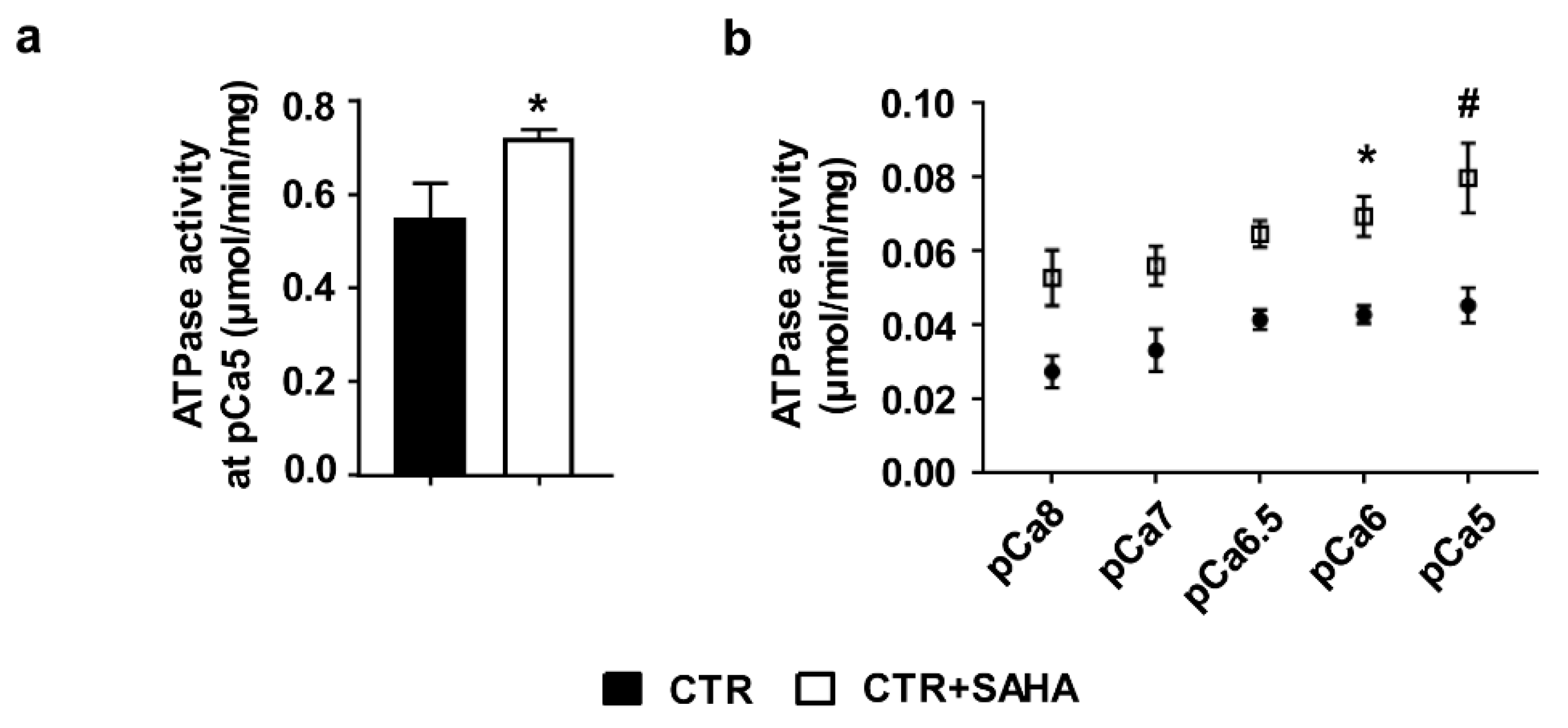
2.1. Effect of SAHA Treatment on SERCA2a Acetylation and Function in Control Rat Cardiomyocytes
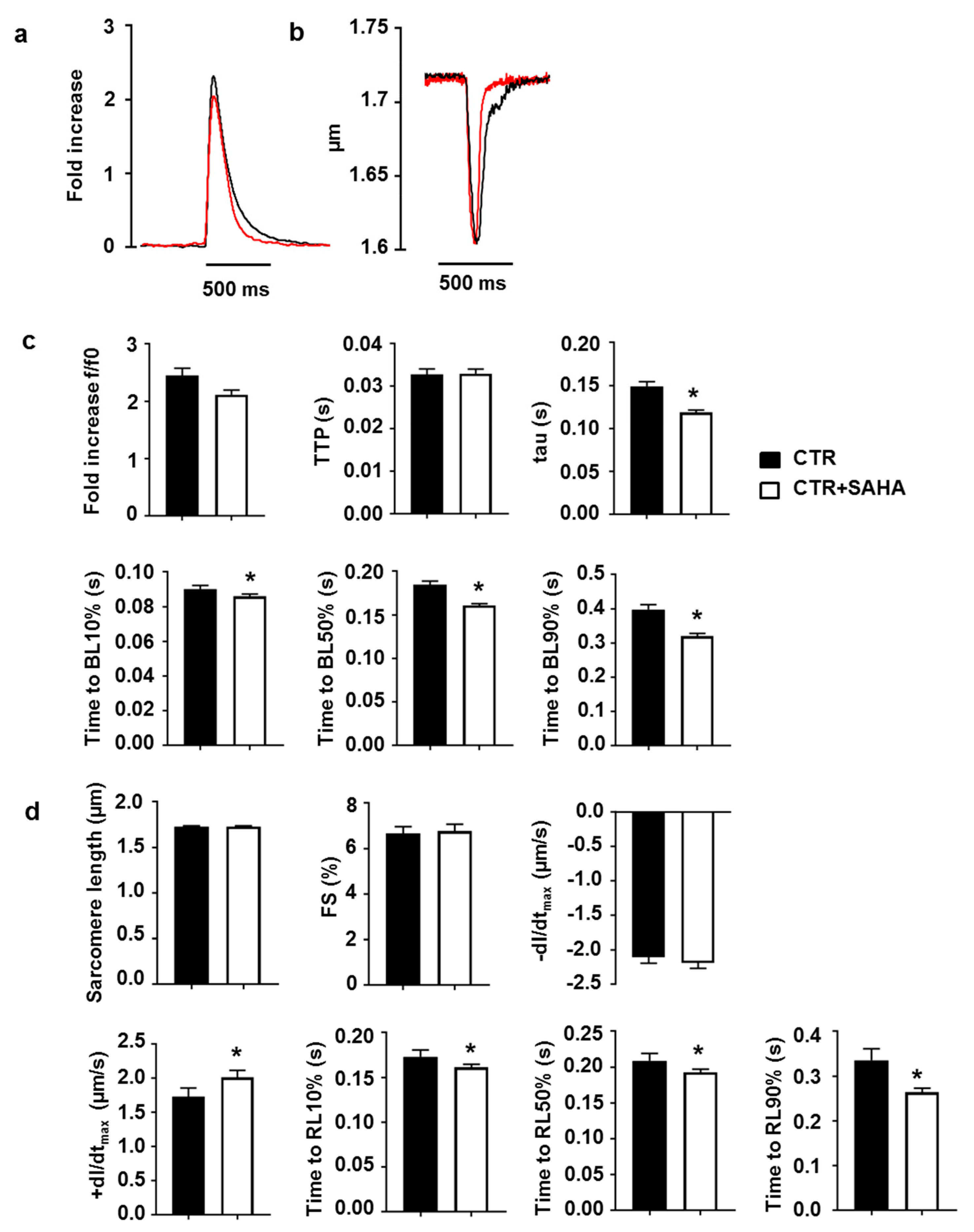
2.2. Effect of SAHA Treatment on Calcium Transients and Cell Mechanics in CMs Isolated from Adult Rat Hearts
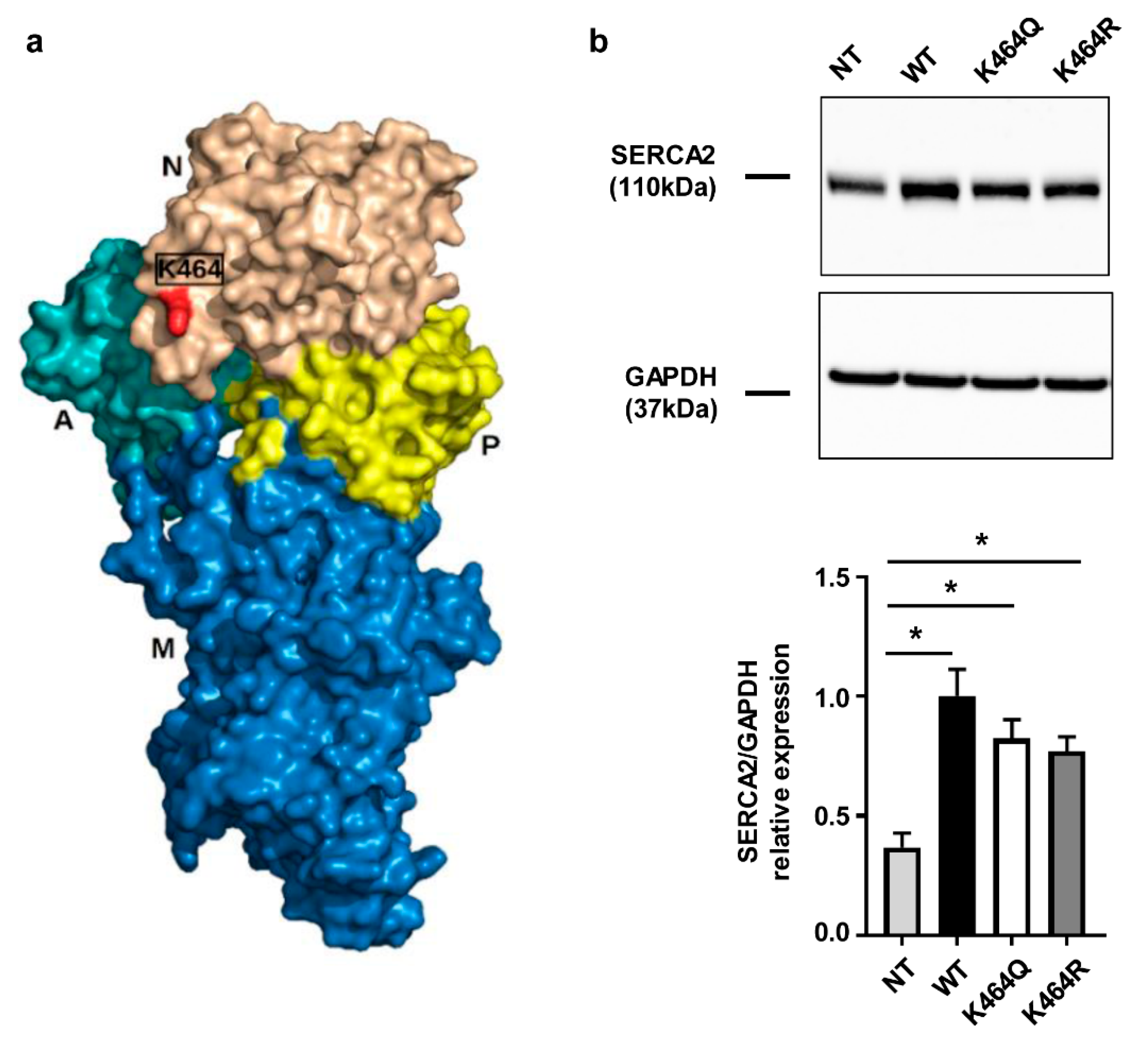
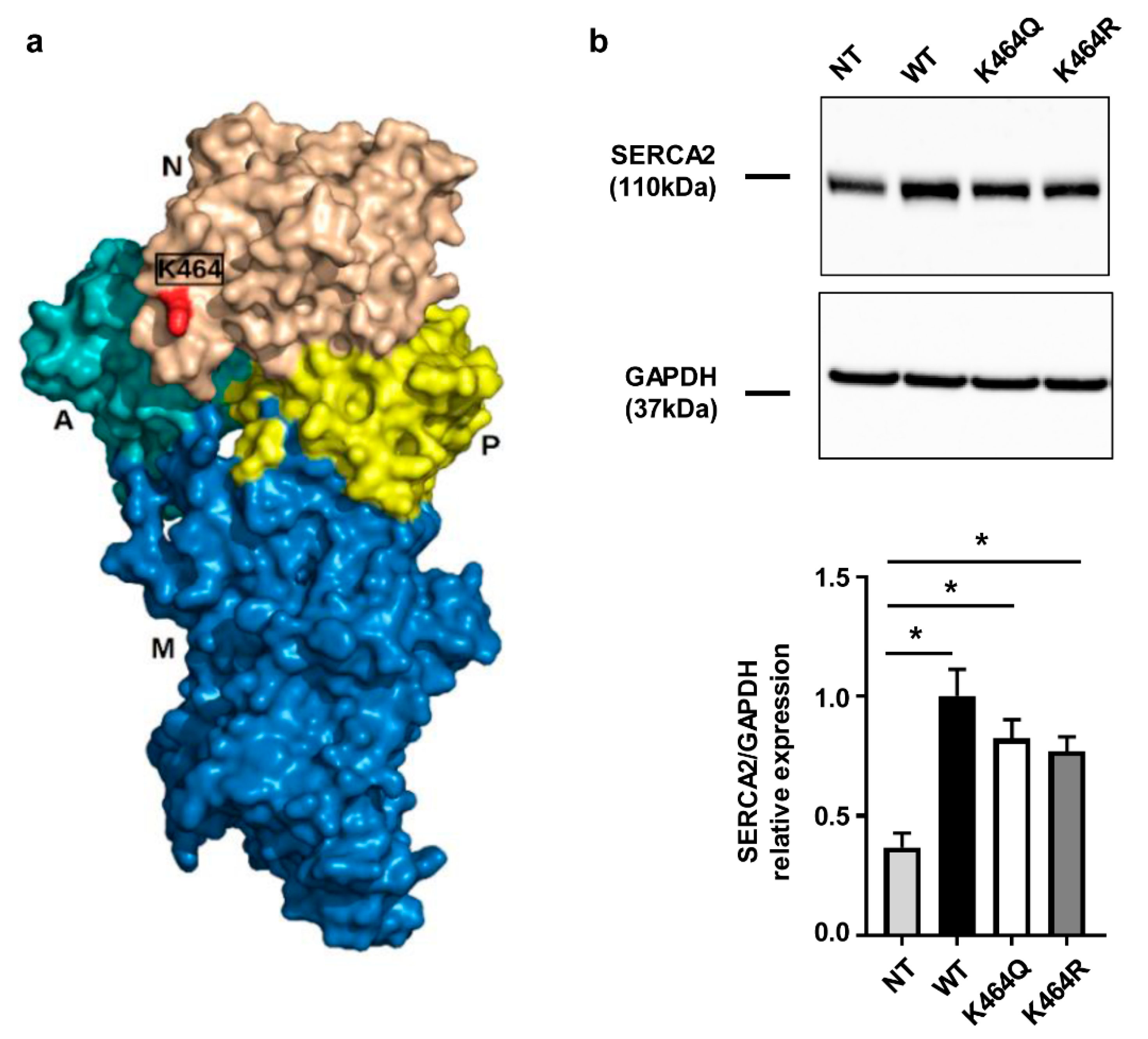
2.3. Nε-Lysine Acetylation Sites of Human SERCA2a
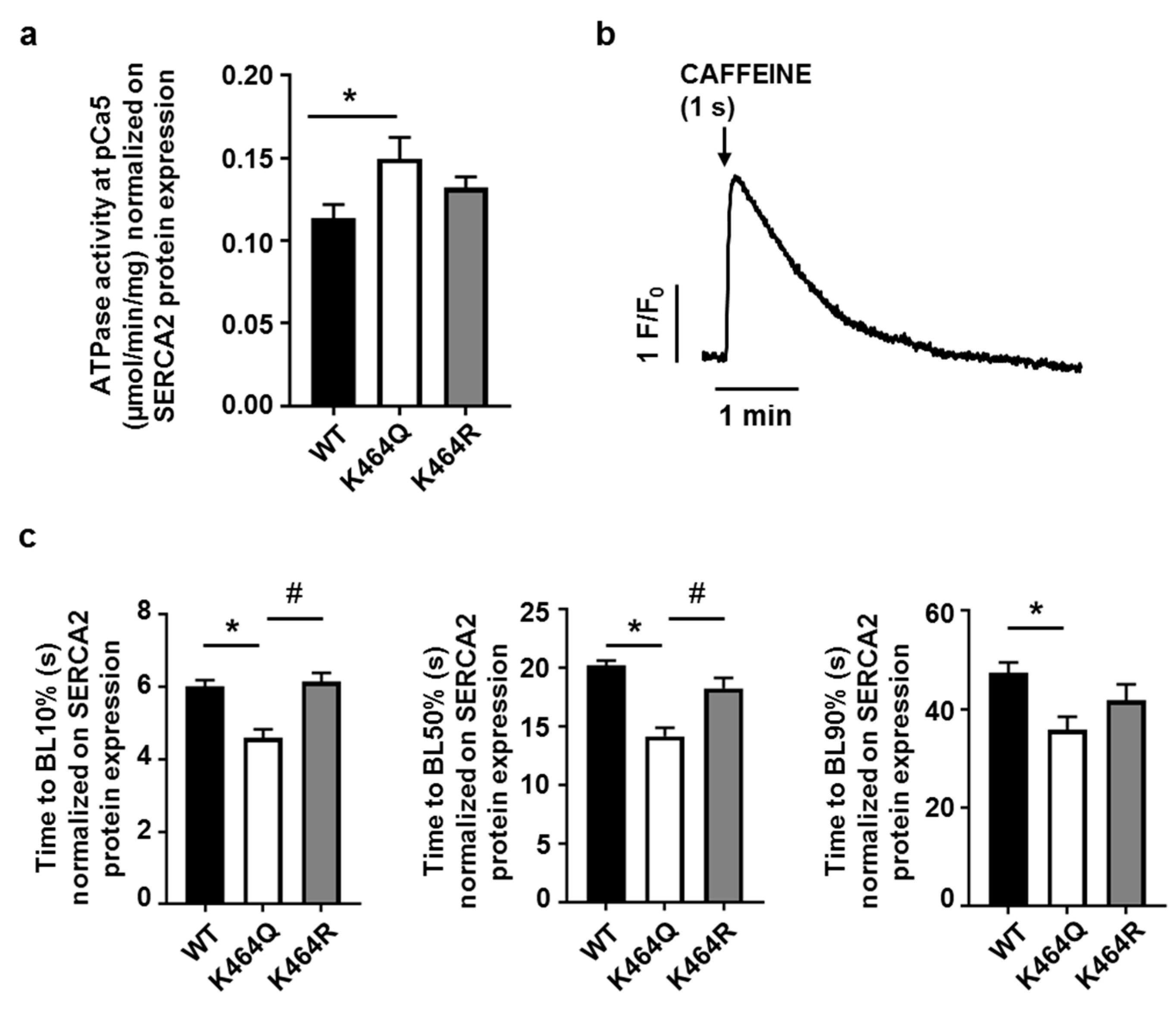
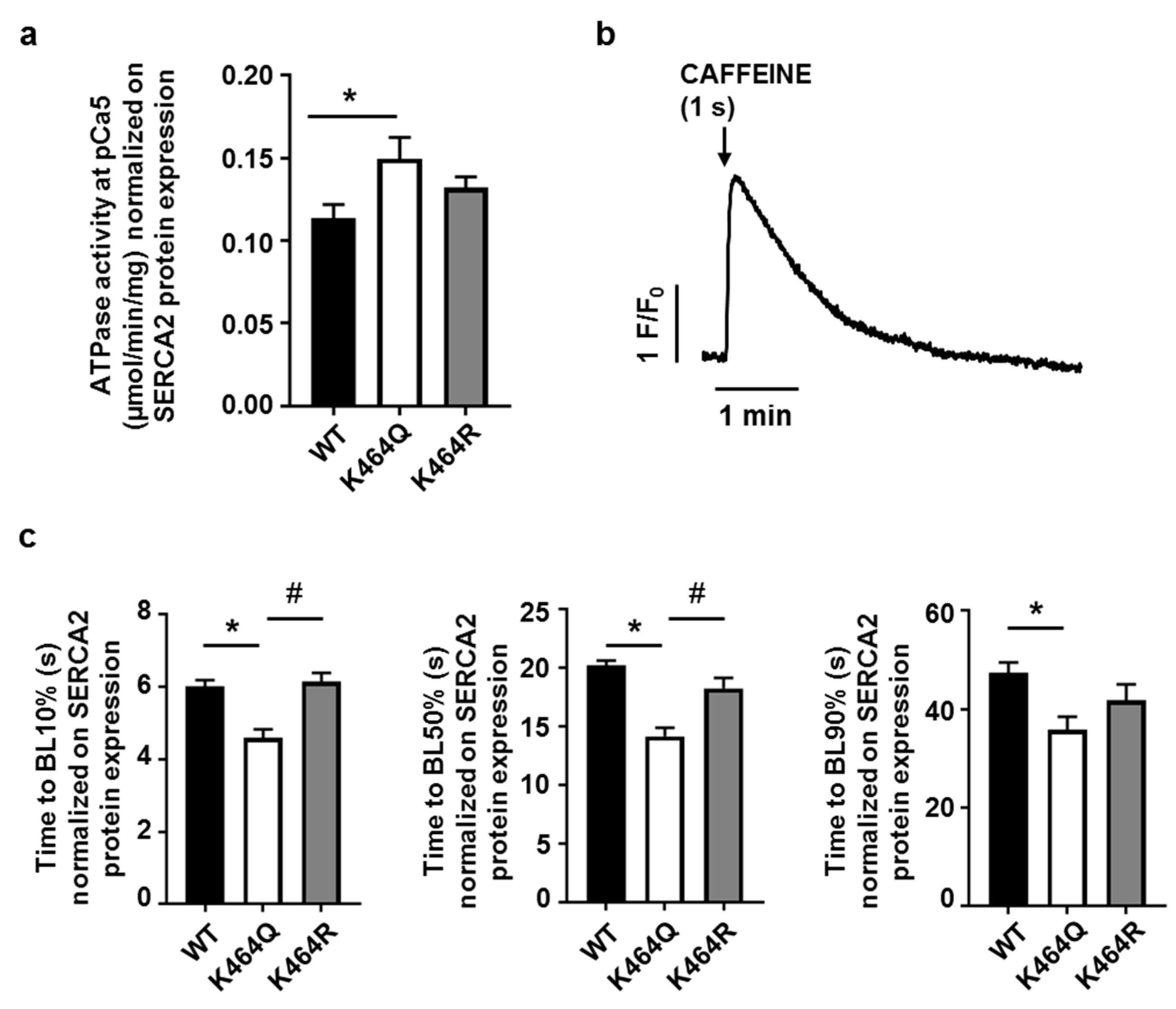
2.4. Effects of K464 Mutation on hSERCA2a ATPase Activity and HEK Calcium Transients
3. Discussion
4. Materials and Methods
4.1. Rat Population
4.2. Rat Cardiomyocyte Isolation
4.3. HL-1 Cardiomyocyte Culture
4.4. Western Blot Analysis
4.5. Immunoprecipitation
4.6. Isolation of the Microsomal Fraction
4.7. ATP/NADH Coupled Assay for Calcium ATPase Activity
4.8. Rat Cardiomyocyte Mechanics and Calcium Transients
4.9. Bioinformatics Analysis
4.10. Mutagenesis
4.1.1. SERCA2 Mutant Calcium Transients
4.1.2. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Toyoshima, C. Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 2008, 476, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Sugita, Y.; Kurzydlowski, K.; De Leon, S.; Tada, M.; Toyoshima, C.; MacLennan, D.H. Sarcolipin regulates sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) by binding to transmembrane helices alone or in association with phospholamban. Proc. Natl. Acad. Sci. USA 2003, 100, 5040–5045. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999, 100, 2308–2311. [Google Scholar] [CrossRef]
- Park, W.J.; Oh, J.G. SERCA2a: A prime target for modulation of cardiac contractility during heart failure. BMB Rep. 2013, 46, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Bhupathy, P.; Babu, G.J. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 2008, 77, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Stammers, A.N.; Susser, S.E.; Hamm, N.C.; Hlynsky, M.W.; Kimber, D.E.; Kehler, D.S.; Duhamel, T.A. The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 2015, 93, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Knyushko, T.V.; Sharov, V.S.; Williams, T.D.; Schoneich, C.; Bigelow, D.J. 3-Nitrotyrosine modification of SERCA2a in the aging heart: A distinct signature of the cellular redox environment. Biochemistry 2005, 44, 13071–13081. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.B.; Liu, T.; Rucker, J.; O’Meally, R.N.; Devine, L.R.; Cole, R.N.; O’Rourke, B. The cardiac acetyl-lysine proteome. PLoS ONE 2013, 8, e67513. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Berni, R.; Rosati, J.; Straino, S.; Vitale, S.; Spallotta, F.; Baruffi, S.; Bocchi, L.; Delucchi, F.; Rossi, S.; et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces cardiac arrhythmias in dystrophic mice. Cardiovasc. Res. 2010, 87, 73–82. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Ferguson, B.S.; Osinska, H.; Bhuiyan, M.S.; James, J.; McKinsey, T.A.; Robbins, J. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, E5178–E5186. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, E.; Testoni, S.; Gentile, A.; Cali, T.; Ottolini, D.; Villa, A.; Brini, M.; Betto, R.; Mascarello, F.; Nissen, P.; et al. Inhibition of ubiquitin proteasome system rescues the defective sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1) protein causing Chianina cattle pseudomyotonia. J. Biol. Chem. 2014, 289, 33073–33082. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J.; Westlin, M.; Burk, S.E.; Shull, G.E.; MacLennan, D.H. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 1992, 267, 14483–14489. [Google Scholar] [PubMed]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Milde, T.; Deubzer, H.E.; Oehme, I.; Witt, R.; Kulozik, A.; Eisenmenger, A.; Abel, U.; Karapanagiotou-Schenkel, I. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klinische Pädiatrie 2012, 224, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Kong, Y.; Tan, W.; May, H.; Battiprolu, P.K.; Pedrozo, Z.; Wang, Z.V.; Morales, C.; Luo, X.; Cho, G.; et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 2014, 129, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Tiffon, C.; Adams, J.; van der Fits, L.; Wen, S.; Townsend, P.; Ganesan, A.; Hodges, E.; Vermeer, M.; Packham, G. The histone deacetylase inhibitors vorinostat and romidepsin downmodulate IL-10 expression in cutaneous T-cell lymphoma cells. Br. J. Pharmacol. 2011, 162, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Simonides, W.S.; van Hardeveld, C. An assay for sarcoplasmic reticulum Ca2+-ATPase activity in muscle homogenates. Anal. Biochem. 1990, 191, 321–331. [Google Scholar] [CrossRef]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Xie, M.; Periyasamy, S.M.; Xie, Z.; Han, C.; Basrur, V.; Mutgi, K.; Fedorov, V.; Malhotra, D.; et al. Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H3003–H3011. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.F.; Bolck, B.; Erdmann, E.; Schwinger, R.H. Sarcoplasmic reticulum Ca2+-ATPase modulates cardiac contraction and relaxation. Cardiovasc. Res. 2003, 57, 20–27. [Google Scholar] [CrossRef]
- Gupta, M.P.; Samant, S.A.; Smith, S.H.; Shroff, S.G. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J. Biol. Chem. 2008, 283, 10135–10146. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Biesiadecki, B.J.; Ziolo, M.T.; Davis, J.P.; Janssen, P.M. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Front. Physiol. 2016, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Chen-Izu, Y.; Shaw, R.M.; Pitt, G.S.; Yarov-Yarovoy, V.; Sack, J.T.; Abriel, H.; Aldrich, R.W.; Belardinelli, L.; Cannell, M.B.; Catterall, W.A.; et al. Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. 2015, 593, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.; Bers, D.M.; Chiamvimonvat, N.; Sato, D. Dynamical effects of calcium-sensitive potassium currents on voltage and calcium alternans. J. Physiol. 2017, 595, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Chen-Izu, Y. Sodium and calcium regulation in cardiac myocytes: From molecules to heart failure and arrhythmia. J. Physiol. 2015, 593, 1327–1329. [Google Scholar] [CrossRef] [PubMed]
- Olesen, C.; Picard, M.; Winther, A.M.; Gyrup, C.; Morth, J.P.; Oxvig, C.; Moller, J.V.; Nissen, P. The structural basis of calcium transport by the calcium pump. Nature 2007, 450, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Tong, X.; Pimentel, D.R.; Weisbrod, R.M.; Trucillo, M.P.; Adachi, T.; Cohen, R.A. Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Du, G.G.; Chen, S.R.; MacLennan, D.H. HEK-293 cells possess a carbachol- and thapsigargin-sensitive intracellular Ca2+ store that is responsive to stop-flow medium changes and insensitive to caffeine and ryanodine. Biochem. J. 1999, 343 Pt 1, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; Haughey, N.J.; Greenway, S.C.; Yacono, P.W.; Golan, D.E.; Geiger, J.D. Expression of ryanodine receptors in human embryonic kidney (HEK293) cells. Biochem. J. 1998, 334 Pt 1, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Sun, H.; Xiao, R.P.; Han, Q. Caffeine induced Ca2+ release and capacitative Ca2+ entry in human embryonic kidney (HEK293) cells. Eur. J. Pharmacol. 2005, 509, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef] [PubMed]
- Blakeslee, W.W.; Wysoczynski, C.L.; Fritz, K.S.; Nyborg, J.K.; Churchill, M.E.; McKinsey, T.A. Class I HDAC inhibition stimulates cardiac protein SUMOylation through a post-translational mechanism. Cell Signal. 2014, 26, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Despa, S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Bostjancic, E.; Zidar, N.; Glavac, D. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: Expression and bioinformatic analysis. BMC Genom. 2012, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lucas, K.J.; Saha, T.T.; Ha, J.; Ling, L.; Kokoza, V.A.; Roy, S.; Raikhel, A.S. MicroRNA-275 targets sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA) to control key functions in the mosquito gut. PLoS Genet. 2017, 13, e1006943. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Shin, S.; Cha, H.J.; Yoon, Y.; Bae, S.; Jung, J.H.; Lee, S.M.; Lee, S.J.; Park, I.C.; Jin, Y.W.; et al. Suberoylanilide hydroxamic acid (SAHA) changes microRNA expression profiles in A549 human non-small cell lung cancer cells. Int. J. Mol. Med. 2009, 24, 45–50. [Google Scholar] [PubMed]
- Yang, H.; Lan, P.; Hou, Z.; Guan, Y.; Zhang, J.; Xu, W.; Tian, Z.; Zhang, C. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br. J. Cancer 2015, 112, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Poddar, S.; Kesharwani, D.; Datta, M. Histone deacetylase inhibition regulates miR-449a levels in skeletal muscle cells. Epigenetics 2016, 11, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, X.; Zheng, H.; Xie, B.; Bidasee, K.R.; Rozanski, G.J. Pro-oxidant effect of transforming growth factor-beta1 mediates contractile dysfunction in rat ventricular myocytes. Cardiovasc. Res. 2008, 77, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Mufti, S.; Wenzel, S.; Euler, G.; Piper, H.M.; Schluter, K.D. Angiotensin II-dependent loss of cardiac function: Mechanisms and pharmacological targets attenuating this effect. J. Cell. Physiol. 2008, 217, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Ammanamanchi, S.; Brattain, M.G. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J. Biol. Chem. 2004, 279, 32620–32625. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-beta1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Santhoshkumar, P.; Reneker, L.W.; Sharma, K.K. Histone deacetylase inhibitors trichostatin A and vorinostat inhibit TGFbeta2-induced lens epithelial-to-mesenchymal cell transition. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar] [CrossRef] [PubMed]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: From important roles in differentiation to possible implications in tumourigenesis. J. Cell. Mol. Med. 2011, 15, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Kahali, S.; Sarcar, B.; Prabhu, A.; Seto, E.; Chinnaiyan, P. Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response. FASEB J. 2012, 26, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Ligeti, L.; Szenczi, O.; Prestia, C.M.; Szabo, C.; Horvath, K.; Marcsek, Z.L.; van Stiphout, R.G.; van Riel, N.A.; Op den Buijs, J.; Van der Vusse, G.J.; et al. Altered calcium handling is an early sign of streptozotocin-induced diabetic cardiomyopathy. Int. J. Mol. Med. 2006, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Kretschmann, B.; Meyer, M.; Holubarsch, C.; Weirich, J.; Posival, H.; Minami, K.; Just, H.; Hasenfuss, G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation 1995, 92, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Dhalla, N.S. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2260–H2270. [Google Scholar] [CrossRef] [PubMed]
- Balke, C.W.; Shorofsky, S.R. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc. Res. 1998, 37, 290–299. [Google Scholar] [CrossRef]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, M.; Pollard, A.E.; Yang, L.; Spitzer, K.W. Beat-to-beat repolarization variability in ventricular myocytes and its suppression by electrical coupling. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H677–H687. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; MacLennan, D.H. Mutation of aspartic acid-351, lysine-352, and lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3314–3318. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, R.; Testoni, S.; Gentile, A.; Damiani, E.; Rossi, M.; Liguori, R.; Drogemuller, C.; Mascarello, F. A defective SERCA1 protein is responsible for congenital pseudomyotonia in Chianina cattle. Am. J. Pathol. 2009, 174, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Kiianitsa, K.; Solinger, J.A.; Heyer, W.D. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal. Biochem. 2003, 321, 266–271. [Google Scholar] [CrossRef]
- Bassani, J.W.; Bassani, R.A.; Bers, D.M. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J. Physiol. 1994, 476, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; Gunawardena, J.; Mann, M. PHOSIDA 2011: The posttranslational modification database. Nucleic Acids Res. 2011, 39, D253–D260. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.K.; Horlick, R.A. Macrophage scavenger receptor confers an adherent phenotype to cells in culture. Biotechniques 1998, 25, 240–244. [Google Scholar] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meraviglia, V.; Bocchi, L.; Sacchetto, R.; Florio, M.C.; Motta, B.M.; Corti, C.; Weichenberger, C.X.; Savi, M.; D’Elia, Y.; Rosato-Siri, M.D.; et al. HDAC Inhibition Improves the Sarcoendoplasmic Reticulum Ca2+-ATPase Activity in Cardiac Myocytes. Int. J. Mol. Sci. 2018, 19, 419. https://doi.org/10.3390/ijms19020419
Meraviglia V, Bocchi L, Sacchetto R, Florio MC, Motta BM, Corti C, Weichenberger CX, Savi M, D’Elia Y, Rosato-Siri MD, et al. HDAC Inhibition Improves the Sarcoendoplasmic Reticulum Ca2+-ATPase Activity in Cardiac Myocytes. International Journal of Molecular Sciences. 2018; 19(2):419. https://doi.org/10.3390/ijms19020419
Chicago/Turabian StyleMeraviglia, Viviana, Leonardo Bocchi, Roberta Sacchetto, Maria Cristina Florio, Benedetta M. Motta, Corrado Corti, Christian X. Weichenberger, Monia Savi, Yuri D’Elia, Marcelo D. Rosato-Siri, and et al. 2018. "HDAC Inhibition Improves the Sarcoendoplasmic Reticulum Ca2+-ATPase Activity in Cardiac Myocytes" International Journal of Molecular Sciences 19, no. 2: 419. https://doi.org/10.3390/ijms19020419