Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD


Centre for Heart Lung Innovation, Department of Medicine, University of British Columbia, Vancouver, BC V6Z1Y6, Canada
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(2), 582; https://doi.org/10.3390/ijms19020582
Submission received: 15 January 2018
/
Revised: 7 February 2018
/
Accepted: 10 February 2018
/
Published: 15 February 2018
(This article belongs to the Special Issue Macrophages in Inflammation)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Lung macrophages (LMs) are essential immune effector cells that are pivotal in both innate and adaptive immune responses to inhaled foreign matter. They either reside within the airways and lung tissues (from early life) or are derived from blood monocytes. Similar to macrophages in other organs and tissues, LMs have natural plasticity and can change phenotype and function depending largely on the microenvironment they reside in. Phenotype changes in lung tissue macrophages have been implicated in chronic inflammatory responses and disease progression of various chronic lung diseases, including Chronic Obstructive Pulmonary Disease (COPD). LMs have a wide variety of functional properties that include phagocytosis (inorganic particulate matter and organic particles, such as viruses/bacteria/fungi), the processing of phagocytosed material, and the production of signaling mediators. Functioning as janitors of the airways, they also play a key role in removing dead and dying cells, as well as cell debris (efferocytic functions). We herein review changes in LM phenotypes during chronic lung disease, focusing on COPD, as well as changes in their functional properties as a result of such shifts. Targeting molecular pathways involved in LM phenotypic shifts could potentially allow for future targeted therapeutic interventions in several diseases, such as COPD.
1. Introduction
The lungs are exposed to the potentially harmful outside environment through the inhalation of gases, particles, suspended toxins, allergens, and pathogens. Accordingly, our immune system has evolved to provide protection against such insults without damaging the lung tissues. Mononuclear phagocytic (MNP) cells line the respiratory tract to process and clear potentially harmful inhaled substances, thereby securing the lung microenvironment. These cells are highly phagocytic in nature, produce large amounts of mediators, and recruit and activate the appropriate immune response to neutralize harmful insults. The majority of these MNP cells are macrophages (~90%) that are distributed throughout the airways, lung tissues, and alveolar tissues. Lung macrophages (LMs) either reside within the lungs or are recruited during lung insult. Resident macrophages self-replicate via a mechanism that is dependent on granulocyte-macrophage colony-stimulating factor (GM-CSF) and colony-stimulating factor-1 (CSF-1) signaling [1]. Therefore, questions have been raised regarding whether most lung tissue and airway macrophages are derived from bone marrow or bone marrow-derived blood monocytes, are recruited into lung tissues and airspaces, and differentiate with limited proliferating capability [2,3]. Recently, self-replicating and proliferating lung tissue-resident macrophages from embryonic [3] and fetal liver origins [4] have been described and characterized in a murine model [5] (Figure 1). These studies have led to the conceptualization of two distinct macrophage populations in the lungs: a tissue-resident population originating from the embryo or fetal liver in early life and a monocyte-derived population originating from hematopoietic stem cells and recruited from the blood. The phenotypic characteristics and functions of these two distinct macrophage populations are an area of active investigation.
2. Lung Macrophages (LM) Phenotypes
LMs have high levels of phenotypic and functional plasticity to adapt and respond to a variety of environmental insults. More than a decade ago, Mills and co-workers proposed a classification of macrophages to capture their phenotypic diversity, suggesting at least two distinct phenotypes, i.e., M1 and M2 macrophages [6]. M1 macrophages produce high levels of pro-inflammatory cytokines, have strong bactericidal properties, produce high levels of reactive nitrogen species and oxygen intermediates, promote a predominantly Th1 inflammatory milieu, and are referred to as “classically activated (pro-inflammatory) macrophages”. Induced by the exposure to Lipopolysaccharide (LPS) and IFN-γ, M1 macrophages secrete large amounts of pro-inflammatory cytokines, including IL-1β, TNF, and IL-12, and strongly express major histocompatibility complex (MHC) II (monocyte–macrophage marker), CD40, CD68, CD80, CD86, and iNOS. In contrast, M2 macrophages have been associated with a more Th2 inflammatory milieu, characterized by allergic responses and protection against parasites. Moreover, M2 macrophages are more immunomodulating in nature, are involved in tissue remodeling, including promoting macrophage efferocytic functions, and are referred to as “alternatively activated macrophages”. Induced by fungi, immunocomplexes, helminths, complement components, apoptotic cells, macrophage colony-stimulating factors, IL-4, and IL-13, M2 macrophages secrete anti-inflammatory cytokines, including IL-10, TGF-β, CCL18, and CCL22 [7], and are generally characterized by their high CD163 and CD206 expression levels [8].
This basic classification by Mills and co-workers has been further expanded by Mantovani and co-workers, dividing M2 macrophages into four different subtypes: M2a, M2b, M2c, and M2d [7]. Further phenotyping by Qian et al. identified tumor-associated macrophages [9] as well as those expressing T cell receptors and CD169. Currently, at least eight different distinct phenotypic populations have been identified [10]. Although the unique roles various M2 macrophages play remain unclear, the notion that macrophage plasticity is determined by the immediate microenvironment has been clearly supported.
3. LM Functional Responses
LMs are essential in regulating innate immune responses in the lungs. They are involved in cell defense against inhaled foreign matter (i.e., ambient irritants, pollutants, bacteria, and viruses), thereby keeping the airways clean. Following phagocytosis, LMs process all foreign materials engulfed and also act as antigen presenting cells, activating the adaptive immune response. These innate and adaptive immune responses are immunologically interdependent with LMs playing a pivotal role in the relationship between both responses. Following the processing of inhaled foreign matter, LMs generate and release mediators that stimulate the bone marrow, promoting the release of monocytes from the marrow, and enhance their recruitment into the lungs where they subsequently differentiate into tissue and airway macrophages [10], further supplementing resident LMs.
LM responses to engulfed foreign matter could somewhat vary depending on the nature of the particles, specifically material size, geometry, and surface characteristics. LMs comfortably phagocytose smaller particles (<10 μm) [11] but frequently collectively cooperate and form giant multinucleated cells to process larger ones (>10 μm) [12]. Champion and Mitragotri [13] showed that particle shape could also impact the ease of LM phagocytosis. In addition, particles with uneven and rougher surfaces promote cell adherence and modulate LM production of pro-inflammatory cytokines and chemokines [14]. Lastly, organic foreign materials, such as microorganisms, require opsonization to promote Fc receptor-mediated phagocytosis and clearance [15] or interact with toll-like receptors that recognize both Gram-positive and Gram-negative bacteria for phagocytosis [16,17].
4. Macrophages in Chronic Obstructive Pulmonary Disease (COPD)
COPD is an inflammatory disease of the lungs that is characterized by air flow limitations that are not fully reversible. It is characterized by clinical symptoms such as cough, sputum production, and shortness of breath with episodes of exacerbation, especially during the late stages of the disease. The disease is progressive in nature and the World Health Organization has estimated that 3.17 million deaths were caused by the COPD in 2015. Although cigarette smoking is still the major cause of COPD in developed countries, biomass exposure is a more significant cause of COPD in developing countries [18]. For both exposure types, LMs play a central role in processing and clearing particles from the lungs. Given the chronicity of these exposures, LMs’ downstream functional responses thereto participate in the development of COPD [19]. A small fraction of COPD are caused by a genetic deficiency in α-1 antitrypsin, but long-term exposure to air pollutants and occupational exposures, such as dust particles, fumes, and chemicals, are more important on a worldwide scale [20]. The significantly increased number of macrophages in the sputum and Bronchoalveolar Lavage Fluid (BALF) of patients with COPD support this notion [21,22]. In COPD, LMs also secrete large amounts of potentially tissue-damaging enzymes, such as elastase, metalloproteinase (MMP)-2, MMP-9, MMP-12, and cathepsin S, in response to foreign particulate matter and microorganism exposure [22,23]. A majority of acute COPD exacerbations are triggered by either viral or bacterial respiratory infections [24] that could lead to bacterial colonization of the airways, subsequently contributing to frequent exacerbations [25]. In addition, continuous exposure to cigarette smoke or biomass markedly depletes intracellular anti-oxidants, such as glutathione, causing excessive oxidative stress, which suppresses LMs’ bacterial phagocytic and efferocytic functions [26]. Therefore, LMs in COPD generate a more pro-inflammatory milieu that could cause tissue damage, as well as defective immune surveillance and protective (phagocytic) functions that collectively contribute to the progression of COPD.
5. LMs and Bacterial Colonization of COPD Airways
The lung airways have their own distinct microbiome as revealed by recent next-generation sequencing technologies, including 16 s RNA gene measurement [20]. Studies in individuals with COPD have revealed alterations in the microbiome that vary with the severity of COPD, acute COPD exacerbations, and use of steroids and/or antibiotics [27]. Changes in the lung microbiome may contribute to the pathogenesis of COPD by influencing inflammatory and/or immune processes in the lungs. Although LMs play a central role in clearing harmful bacteria from the lungs, this function deteriorates as the disease progresses. Berenson et al. reported that the severity of COPD (FEV 1% predicted) correlated with impaired LM phagocytosis for nontypeable Haemophilus influenzae (NTHi) and Moraxella catarrhalis [28]. Moreover, studies using clone library analysis of the 16 s RNA gene sequence have reported that H. influenzae and M. catarrhalis were more frequently identified as causative bacteria for pneumonia and/or exacerbations of COPD as well as disease progression [29]. Similarly, Streptococcus pneumoniae, which also causes acute COPD exacerbations, had been associated with the progression of emphysema possibly by inducing LMs to produce MMP-12 [30]. Therefore, the defective phagocytic and efferocytic functions of LMs in COPD could contribute to the colonization of the airways with various bacteria, specifically those known to cause acute exacerbations and pneumonia during COPD. Consequently, these infections have been related to the progression of COPD [29].
A pivotal function of LMs is to remove and clear cell debris and dead or damaged cells following an inflammatory insult to the lungs. Such a process, which is termed efferocytosis, becomes defective in subjects with COPD [31]. Accordingly, sputum and airway neutrophils are elevated in most subjects with COPD and further increase during acute COPD exacerbations [32]. The defective clearance of these recruited neutrophils allows for the accumulation of necrotic neutrophils that indiscriminately release toxic granule proteins containing neutrophil elastase, a protease that has been associated with tissue damage and COPD progression [33]. The recognition of activated neutrophil extracellular trap-like structures containing their DNA and granule proteins, such as histones and neutrophil elastase [34], is imperative for the proper clearance of cell debris following an inflammatory insult to the lungs. Neutrophil elastase degrades extracellular matrix components as well as lung elastic fibers related to the development of pulmonary emphysema, and promotes the release of mucin via an epidermal growth factor receptor-dependent mechanism [35]. Considering that LMs are the primary “janitors” of the lungs, dysfunctional processing and clearance of apoptotic and necrotic cells and cell debris could clearly contribute to ongoing lung tissue inflammation in subjects with COPD, even long after they stop smoking [36].
6. Macrophage Phenotypes in COPD
The role of distinct macrophage phenotypes (M1 versus M2) in COPD is unclear. Environmental exposures causing COPD produce either an acute or chronic insult resulting in an inflammatory response in the lung tissues that promotes both M1 (pro-inflammatory) and M2 (reparative) macrophages. An example would be the simultaneous elevation of iNOS (an M1 marker) and arginase (an M2 marker) activity in COPD tissue [37]. Furthermore, non-polarized macrophages that are negative for both M1 and M2 markers or dual positive for both markers have been reported by several investigators [37]. Non-polarized macrophages are more frequently distributed in normal lung tissues, while dual positive (both M1 and M2 marker-positive) macrophages are more frequently distributed in lung tissues of subjects with severe COPD [37]. The presence of COPD suggests that these macrophages have been recently recruited (not resident) and both classically (M1) and alternatively (M2) activated through the expression of markers for both phenotypes. Both M1 and M2 macrophages have been reported in smokers with and without COPD and other lung diseases, such as sarcoidosis [38]. Nonetheless, the plasticity of macrophages to change phenotypes largely depends on their microenvironment [39] (Figure 2). Using a monocyte cell line (THP-1 cells), Genin and co-workers generated macrophages through incubation with phorbol 12-myristate 13-acetate. The macrophages they obtained did not possess any of the M1 or M2 activation markers (M0 macrophages). By incubating these M0 macrophages with specific phenotypic mediators (see Figure 2), they were able to obtain differentiated and polarized M1 and M2 macrophages [40]. The complexity of the COPD microenvironment is such that four distinct phenotypes of macrophages can be clearly identified (Figure 3A): non-polarized macrophages (both M1 and M2 marker-negative), M1-skewed macrophages (strong M1 marker-positive), M2-skewed macrophages (strong M2 marker-positive), and hybrid macrophages (both M1 and M2 marker-positive or dual positive). Little is known about the distribution of these different phenotypes of macrophages in the tissues and airspaces in the COPD lung. Recently, Eapen and co-workers showed that non-polarized macrophages were present in both small airway tissues and luminal airspaces, whereas M1 macrophages predominate in small airways and M2 macrophages predominate in luminal areas in COPD lung tissues compared to normal control [41] (Figure 3B). Owing to chronic and ongoing insult (cigarette smoke and colonization/infection), individuals with COPD experience constant changes to their lung environment, potentially influencing macrophage phenotype with time. Accordingly, elucidating the origin and functional properties of these four LM phenotypes could pave the way for the development of more macrophage-specific therapeutic interventions for COPD.
7. Phagocytic and Efferocytic Functions of Macrophages in COPD
Inhaled foreign substances, such as bacteria, fungi, viruses, pollen, and air pollutants, are processed and cleared from the lungs to maintain homeostasis. During COPD, the total number of airway macrophages (BAL) are significantly increased, while phagocytosis and elimination of microorganisms and apoptotic cells are paradoxically impaired [42], suggesting defective functional properties of the macrophages. Several investigators have reported impairments in LMs’ ability to phagocytose fungi, bacteria, and air pollution (in a study using latex beads) (compared to normal controls) [28], while patients with COPD also displayed impaired LM efferocytosis [43]. Furthermore, peripheral blood monocytes from patients with COPD were significantly less effective in killing Candida albicans than those from control subjects [44,45], suggesting the presence of these defects even before recruitment into lung tissues. In a rat model of COPD, macrophage phagocytosis of Aspergillus had been impaired [46]. Moreover, LMs from donors with COPD had a greater impairment in phagocytic activity against NTHi compared to those from donors without COPD [47,48]. Macrophages derived from blood monocytes of patients with COPD have reduced phagocytic activity against H. influenzae and S. pneumoniae [49]. However, COPD macrophages showed no defect in the non-specific phagocytosis of latex beads [43,49], suggesting a defect in receptor- or opsonization-mediated phagocytosis. This notion has been supported by studies using rhinovirus exposure to impair LM phagocytosis and immune responses to bacterial products [50]. Clearly, the mechanisms of LM phagocytic dysfunction remain unclear and need further study.
Efferocytosis, a process by which dead and apoptotic cells are removed from the body, is an essential function for maintaining a healthy lung microenvironment. Various studies have reported impairments in the efferocytic function of LMs among patients with COPD [42]. A significantly greater impairment in efferocytosis had been reported in patients with non-eosinophilic asthma or COPD compared to those with eosinophilic asthma [32]. Although the underlying mechanism for this disorder has yet to be fully elucidated, several hypotheses have been proposed. A cigarette-smoking model revealed that apoptotic cell clearance had been impaired through oxidant-dependent activation of RhoA [51]. Furthermore, galectin-3, an S-type lectin known to regulate the phenotype and function of macrophages, is significantly decreased in the BAL of patients with COPD [52]. Lastly, Hodge et al. reported that reduced LM efferocytosis in patients with COPD was associated with a reduced expression of M2 mannose receptors (CD206) and several other key macrophage recognition molecules [42], suggesting a direct link between macrophage functional properties and phenotype. Defective efferocytosis allows for cytotoxic products of dead or dying cells to be released, possibly leading to lung tissue damage and COPD progression.
8. Therapeutic Targeting of Macrophages in COPD
Currently, no pharmacological COPD intervention has been shown to significantly slow the decline in FEV1 over time. Although bronchodilators have been the primary treatment option for airflow limitations typical in COPD [24], they predominantly address the symptoms and not the underlying immune pathogenesis of the disease. Novel treatments target specific molecular pathways thought to be pivotal in the pathogenesis of the disease, such as small molecule inhibitors with anti-inflammatory activity against p38 mitogen-activated protein kinases, phosphatidyl-inositol-3 kinase, and Rho kinase [53].
Hodge et al. reported that azithromycin increases the phagocytosis of apoptotic bronchial epithelial cells [54] and improves the phagocytosis of bacteria through both alveolar- and monocyte-derived macrophages in patients with COPD [55,56]. Macrolides with diminished antibiotic activity (non-antibiotic macrolides) significantly improved the phagocytosis of apoptotic cells and NTHi, an approach that could potentially reduce airway inflammation in patients with chronic inflammatory lung conditions, such as COPD and bronchiectasis [57]. In a recent study, azithromycin 250 mg daily for 8 weeks altered both lung microbiota and metabolome in patients with COPD [58]. According to these reports, macrolides such as azithromycin might contribute to COPD treatment by changing the lung microbiome via macrophage phagocytic function.
Several epidemiological studies have shown that inhaled corticosteroids increase the incidence of pneumonia [59,60], tuberculosis, and upper respiratory tract infections [61] in patients with COPD. Mouse models have shown that fluticasone treatment in the presence of apoptotic cells significantly reduced in vitro LM killing of pneumococci in part by delaying phagolysosome acidification without affecting the production of reactive oxygen or nitrogen species [62]. Inhaled fluticasone propionate has also been shown to impair pulmonary clearance of Klebsiella pneumoniae [63]. Treatment with lovastatin [64], low-dose oxygen [65], and the acute phase reactant α-1 antitrypsin [66] increased the efferocytosis function of LMs in a mouse model. Moreover, several studies have shown that statins beneficially impact innate immune responses in the lungs, including in subjects with COPD [67]. They also reduce airway macrophages and pro-inflammatory mediators, such as IL-17, and increase anti-inflammatory mediators, such as IL-10 [68].
Collectively, these studies suggest that altering the recruitment and functional properties of LMs could potentially reduce ongoing inflammatory responses in lung tissues of subjects with COPD.
Macrophage efferocytosis of eosinophils is impaired in patients with COPD, the degree of which is related to the severity and frequency of COPD exacerbations [69]. In human alveolar macrophages from patients with COPD [70] and in LMs of mice exposed to cigarette smoke [71], d-series resolvins reduced the levels of pro-inflammatory cytokines produced and enhanced phagocytic function, suggesting that cigarette smoke exposure impairs efferocytosis via the inhibition of the HDAC/Rac/CD9 pathways [72]. Phosphodiesterase (PDE) inhibitors, such as aminophylline/theophylline, are effective in restoring efferocytosis impairment in LMs, while recent studies using roflumilast [73], a newer PDE-4 inhibitor, revealed alterations in the LM phenotype to a more reparative M2 type [74]. These studies underscore the known anti-inflammatory properties of PDE inhibitors, suggesting that such effects are produced via impacting LM function by promoting their anti-inflammatory or reparative functions [75].
In a mouse model, vitamin D-deficient mice exhibited an accelerated decline in lung function and a greatly impaired ex vivo phagocytic and oxidative burst function of alveolar macrophages after cigarette smoke exposure compared to control mice [76]. Accordingly, the administration of bisphosphonate alendronate via aerosol inhalation in such mice not only ameliorated elastase-induced emphysema by inhibiting macrophage migratory and phagocytic activities but also blunted the inflammatory response of alveolar macrophages by inhibiting nuclear factor-κB signaling [77]. Lastly, oxidative stress reduces the levels of macrophage phagocytic activity in COPD [78]. Oxidized phospholipids generated through chronic cigarette smoke exposure play a crucial role in inhibiting the phagocytic function of LMs, thus impairing the innate pulmonary anti-bacterial defenses in mice exposed to cigarette smoke [79]. Therapeutic approaches that augment pulmonary antioxidant defenses could be beneficial for reducing the oxidative stress-driven impairment in LM phagocytosis among smokers and patients with COPD [80,81].
9. Conclusions
LMs are essential innate immune effector cells that elicit and control inflammatory responses in the lungs after exposure to environmental pollutants, including cigarette smoke: the main environmental risk factor for COPD. Although LMs are pivotal for processing and removing inhaled irritants from the lungs, they could also inflict damage to lung tissues in the process by promoting a dysregulated inflammatory response, which may in turn lead to dysfunctional tissue repair and a persistent state of chronic low-grade inflammation in the lungs. Despite being poorly understood, the immune pathogenic mechanisms by which COPD develops and progresses need to be elucidated in order to manage and treat the disease. Given that LMs are essential effector cells in this process, we herein describe the characteristics and behavior of macrophages in COPD. Studies that unravel the mechanisms promoting macrophage anti-inflammatory and reparative functions could contribute to the development of more targeted therapeutic interventions to reduce the destructive inflammatory response induced by cigarette smoke and environmental exposures that eventually lead to COPD.
Acknowledgments
The authors declare no conflicts of interest in association with the present study. This work was funded by the British Columbia Lung Association and Stephan F. van Eeden is the CIHR/GSK Professor in Chronic Obstructive Pulmonary Disease.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the TH1/TH2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ishii, H.; Hogg, J.C.; Shih, C.H.; Yatera, K.; Vincent, R.; van Eeden, S.F. Particulate matter air pollution stimulates monocyte release from the bone marrow. Am. J. Respir. Crit. Care Med. 2004, 170, 891–897. [Google Scholar] [CrossRef] [PubMed]
- van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef] [PubMed]
- Refai, A.K.; Textor, M.; Brunette, D.M.; Waterfield, J.D. Effect of titanium surface topography on macrophage activation and secretion of proinflammatory cytokines and chemokines. J. Biomed. Mater. Res. A 2004, 70, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Lee, M.E.; Iannelli, F.; Pozzi, G.; Mitchell, T.J.; Read, R.C.; Dockrell, D.H. Streptococcus pneumoniae-associated human macrophage apoptosis after bacterial internalization via complement and Fcgamma receptors correlates with intracellular bacterial load. J. Infect. Dis. 2003, 188, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Biondo, C.; Mancuso, G.; Beninati, C.; Iaria, C.; Romeo, O.; Cascio, A.; Teti, G. The role of endosomal toll-like receptors in bacterial recognition. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1506–1512. [Google Scholar] [PubMed]
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R.; Committee on Nonsmoking COPD, Environmental and Occupational Health Assembly. An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Tuder, R.M. Apoptosis in the lung: Induction, clearance and detection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L601–L611. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.E.; Culpitt, S.V.; DeMatos, C.; Donnelly, L.; Smith, M.; Wiggins, J.; Barnes, P.J. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2002, 26, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Nakamura, H.; Owen, C.A.; Yoshida, S.; Tsuduki, K.; Chubachi, S.; Shirahata, T.; Mashimo, S.; Nakamura, M.; Takahashi, S.; et al. Plasma Cathepsin S and Cathepsin S/Cystatin C ratios are potential biomarkers for COPD. Dis. Markers 2016, 2016, 4093870. [Google Scholar] [CrossRef] [PubMed]
- Global Strategy for the diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lungo Disease (GOLD). 2017. Available online: http://goldcopd.org (accessed on 20 December 2017).
- Zakharkina, T.; Koczulla, A.R.; Mardanova, O.; Hattesohl, A.; Bals, R. Detection of microorganisms in exhaled breath condensate during acute exacerbations of COPD. Respirology 2011, 16, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.E.; Barnes, P.J. Defective phagocytosis in airways disease. Chest 2012, 141, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.J.; Sethi, S. COPD and the microbiome. Respirology 2016, 21, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Berenson, C.S.; Kruzel, R.L.; Eberhardt, E.; Sethi, S. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J. Infect. Dis. 2013, 208, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Yamasaki, K.; Yatera, K.; Akata, K.; Noguchi, S.; Kawanami, T.; Fukuda, K.; Kido, T.; Ishimoto, H.; Mukae, H. Bacteriological incidence in pneumonia patients with pulmonary emphysema: A bacterial floral analysis using the 16S ribosomal RNA gene in bronchoalveolar lavage fluid. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Ishii, M.; Namkoong, H.; Hegab, A.E.; Asami, T.; Yagi, K.; Sasaki, M.; Haraguchi, M.; Sato, M.; Kameyama, N.; et al. Pneumococcal infection aggravates elastase-induced emphysema via matrix metalloproteinase 12 overexpression. J. Infect. Dis. 2016, 213, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Spooner, G.; Rahman, I.; Rossi, A.G. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem. Biophys. Res. Commun. 2004, 318, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Gibson, P.G.; Yang, I.A.; Upham, J.; James, A.; Reynolds, P.N.; Hodge, S. AMAZES Study Research Group. Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin. Exp. Allergy 2013, 43, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of proteases in chronic obstructive pulmonary disease. Front. Pharmacol. 2017, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Carroll, T.P.; Greene, C.M.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Effect of pro-inflammatory stimuli on mucin expression and inhibition by secretory leucoprotease inhibitor. Cell. Microbiol. 2007, 9, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Osinska, I.; Wolosz, D.; Domagala-Kulawik, J. Association between M1 and M2 macrophages in bronchoalveolar lavage fluid and tobacco smoking in patients with sarcoidosis. Pol. Arch. Med. Wewn. 2014, 124, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Thomas, A.C.; Lenz, L.L.; Munder, M. Macrophage: SHIP of immunity. Front. Immunol. 2014, 5, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 13392. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2007, 37, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003, 81, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Vecchiarelli, A.; Dottorini, M.; Puliti, M.; Todisco, T.; Cenci, E.; Bistoni, F. Defective candidacidal activity of alveolar macrophages and peripheral blood monocytes from patients with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1991, 143, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; D’Adda, D.; Falchi, M.; Dall’Asta, L. The macrophagic activity of patients affected by pneumonia or chronic obstructive pulmonary disease. Int. J. Tissue React. 1996, 18, 109–114. [Google Scholar] [PubMed]
- Wu, Y.; Xu, H.; Li, L.; Yuan, W.; Zhang, D.; Huang, W. Susceptibility to Aspergillus infections in rats with chronic obstructive pulmonary disease via deficiency function of alveolar macrophages and impaired activation of TLR2. Inflammation 2016, 39, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Berenson, C.S.; Garlipp, M.A.; Grove, L.J.; Maloney, J.; Sethi, S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J. Infect. Dis. 2006, 194, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.; Ahern, J.; Carroll, A.; Hodge, G.; Pizzutto, S.; Jersmann, H.; Reynolds, P.; Hodge, S. A small volume technique to examine and compare alveolar macrophage phagocytosis of apoptotic cells and non typeable Haemophilus influenzae (NTHi). J. Immunol. Methods 2016, 429, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.E.; Finney-Hayward, T.K.; Quint, J.K.; Thomas, C.M.; Tudhope, S.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. Defective macrophage phagocytosis of bacteria in COPD. Eur. Respir. J. 2010, 35, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Oliver, B.G.; Lim, S.; Wark, P.; Laza-Stanca, V.; King, N.; Black, J.L.; Burgess, J.K.; Roth, M.; Johnston, S.L. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax 2008, 63, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Richens, T.R.; Linderman, D.J.; Horstmann, S.A.; Lambert, C.; Xiao, Y.Q.; Keith, R.L.; Boe, D.M.; Morimoto, K.; Bowler, R.P.; Day, B.J.; et al. Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am. J. Respir. Crit. Care Med. 2009, 179, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Mukaro, V.R.; Bylund, J.; Hodge, G.; Holmes, M.; Jersmann, H.; Reynolds, P.N.; Hodge, S. Lectins offer new perspectives in the development of macrophage-targeted therapies for COPD/emphysema. PLoS ONE 2013, 8, e56147. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.A.; Belchamber, K.B.; Chana, K.K.; Budd, R.C.; Donaldson, G.; Wedzicha, J.A.; Brightling, C.E.; Kilty, I.; Donnelly, L.E.; Barnes, P.J.; et al. Differential effects of p38, MAPK, PI3K or Rho kinase inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS ONE 2016, 11, e0163139. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Brozyna, S.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur. Respir. J. 2006, 28, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Jersmann, H.; Matthews, G.; Ahern, J.; Holmes, M.; Reynolds, P.N. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 178, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Reynolds, P.N. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology 2012, 17, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Tran, H.B.; Hamon, R.; Roscioli, E.; Hodge, G.; Jersmann, H.; Ween, M.; Reynolds, P.N.; Yeung, A.; Treiberg, J.; et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L678–L687. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.N.; Clemente, J.C.; Wu, B.G.; Wikoff, W.R.; Gao, Z.; Li, Y.; Ko, J.P.; Rom, W.N.; Blaser, M.J.; Weiden, M.D. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax 2017, 72, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Amin, A.V.; Loke, Y.K. Long-term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: A meta-analysis. Arch. Intern. Med. 2009, 169, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Tashkin, D.; Zhang, X.; Radner, F.; Sjobring, U.; Thoren, A.; Calverley, P.M.; Rennard, S.I. Budesonide and the risk of pneumonia: A meta-analysis of individual patient data. Lancet 2009, 374, 712–719. [Google Scholar] [CrossRef]
- Yang, M.; Chen, H.; Zhang, Y.; Du, Y.; Xu, Y.; Jiang, P.; Xu, Z. Long-term use of inhaled corticosteroids and risk of upper respiratory tract infection in chronic obstructive pulmonary disease: A meta-analysis. Inhal. Toxicol. 2017, 29, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Stolberg, V.R.; McCubbrey, A.L.; Freeman, C.M.; Brown, J.P.; Crudgington, S.W.; Taitano, S.H.; Saxton, B.L.; Mancuso, P.; Curtis, J.L. Glucocorticoid-augmented efferocytosis inhibits pulmonary pneumococcal clearance in mice by reducing alveolar macrophage bactericidal function. J. Immunol. 2015, 195, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.M.; Morrison, R.L.; D’Souza, A.; Teng, X.S.; Happel, K.I. Inhaled fluticasone propionate impairs pulmonary clearance of Klebsiella pneumoniae in mice. Respir. Res. 2012, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [PubMed]
- Bain, W.G.; Tripathi, A.; Mandke, P.; Gans, J.H.; D’Alessio, F.R.; Sidhaye, V.K.; Aggarwal, N.R. Low-dose oxygen enhances macrophage-derived bacterial clearance following cigarette smoke exposure. J. Immunol. Res. 2016, 2016, 1280347. [Google Scholar] [CrossRef] [PubMed]
- Serban, K.A.; Petrusca, D.N.; Mikosz, A.; Poirier, C.; Lockett, A.D.; Saint, L.; Justice, M.J.; Twigg, H.L., 3rd; Campos, M.A.; Petrache, I. Alpha-1 antitrypsin supplementation improves alveolar macrophages efferocytosis and phagocytosis following cigarette smoke exposure. PLoS ONE 2017, 12, e0176073. [Google Scholar] [CrossRef] [PubMed]
- Walton, G.M.; Stockley, J.A.; Griffiths, D.; Sadhra, C.S.; Purvis, T.; Sapey, E. Repurposing treatments to enhance innate immunity. Can statins improve neutrophil functions and clinical outcomes in COPD? J. Clin. Med. 2016, 5, E89. [Google Scholar] [CrossRef] [PubMed]
- Maneechotesuwan, K.; Wongkajornsilp, A.; Adcock, I.M.; Barnes, P.J. Simvastatin suppresses airway IL-17 and upregulates IL-10 in patients with stable COPD. Chest 2015, 148, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Eltboli, O.; Bafadhel, M.; Hollins, F.; Wright, A.; Hargadon, B.; Kulkarni, N.; Brightling, C. COPD exacerbation severity and frequency is associated with impaired macrophage efferocytosis of eosinophils. BMC Pulm. Med. 2014, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Thatcher, T.H.; Kottmann, R.M.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Sime, P.J.; Phipps, R.P. Resolvins attenuate inflammation and promote resolution in cigarette smoke-exposed human macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L888–L901. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.M.; Sapinoro, R.E.; Thatcher, T.H.; Croasdell, A.; Levy, E.P.; Fulton, R.A.; Olsen, K.C.; Pollock, S.J.; Serhan, C.N.; Phipps, R.P.; et al. A novel anti-inflammatory and pro-resolving role for resolvin D1 in acute cigarette smoke-induced lung inflammation. PLoS ONE 2013, 8, e58258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, N.; Matsumoto, K.; Fukuyama, S.; Asai, Y.; Kitajima, H.; Seki, N.; Matsunaga, Y.; Kan, O.K.; Moriwaki, A.; Morimoto, K.; et al. Cigarette smoke impairs phagocytosis of apoptotic neutrophils by alveolar macrophages via inhibition of the histone deacetylase/Rac/CD9 pathways. Int. Immunol. 2013, 25, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Martorana, P.A.; Lunghi, B.; Lucattelli, M.; De Cunto, G.; Beume, R.; Lungarella, G. Effect of roflumilast on inflammatory cells in the lungs of cigarette smoke-exposed mice. BMC Pulm. Med. 2008, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Vrancic, M.; Banjanac, M.; Nujic, K.; Bosnar, M.; Murati, T.; Munic, V.; Stupin Polancec, D.; Belamaric, D.; Parnham, M.J.; Erakovic Haber, V. Azithromycin distinctively modulates classical activation of human monocytes in vitro. Br. J. Pharmacol. 2012, 165, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Buenestado, A.; Chaumais, M.C.; Grassin-Delyle, S.; Risse, P.A.; Naline, E.; Longchampt, E.; Tenor, H.; Devillier, P. Roflumilast inhibits lipopolysaccharide-induced tumor necrosis factor-alpha and chemokine production by human lung parenchyma. PLoS ONE 2013, 8, e74640. [Google Scholar] [CrossRef] [PubMed]
- Heulens, N.; Korf, H.; Cielen, N.; De Smidt, E.; Maes, K.; Gysemans, C.; Verbeken, E.; Gayan-Ramirez, G.; Mathieu, C.; Janssens, W. Vitamin D deficiency exacerbates COPD-like characteristics in the lungs of cigarette smoke-exposed mice. Respir. Res. 2015, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nishimura, S.; Ogata, F.; Masubuchi, H.; Hara, K.; Yamaguchi, K.; Aoki, F.; Suga, T.; Nagai, R.; et al. Alendronate inhalation ameliorates elastase-induced pulmonary emphysema in mice by induction of apoptosis of alveolar macrophages. Nat. Commun. 2015, 6, 6332. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.B.; Ahern, J.; Hodge, G.; Holt, P.; Dean, M.M.; Reynolds, P.N.; Hodge, S. Oxidative stress decreases functional airway mannose binding lectin in COPD. PLoS ONE 2014, 9, e98571. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Gang, X.; Kim, J.H.; Sussan, T.E.; Witztum, J.L.; Biswal, S. Oxidized phospholipids impair pulmonary antibacterial defenses: Evidence in mice exposed to cigarette smoke. Biochem. Biophys. Res. Commun. 2012, 426, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Aaron, S.D.; Vandemheen, K.L.; Maltais, F.; Field, S.K.; Sin, D.D.; Bourbeau, J.; Marciniuk, D.D.; FitzGerald, J.M.; Nair, P.; Mallick, R. TNF alpha antagonists for acute exacerbations of COPD: A randomised double-blind controlled trial. Thorax 2013, 68, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Ernst, P.; Hudson, M. TNF-alpha antagonists and the prevention of hospitalisation for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2008, 21, 234–238. [Google Scholar] [CrossRef] [PubMed]
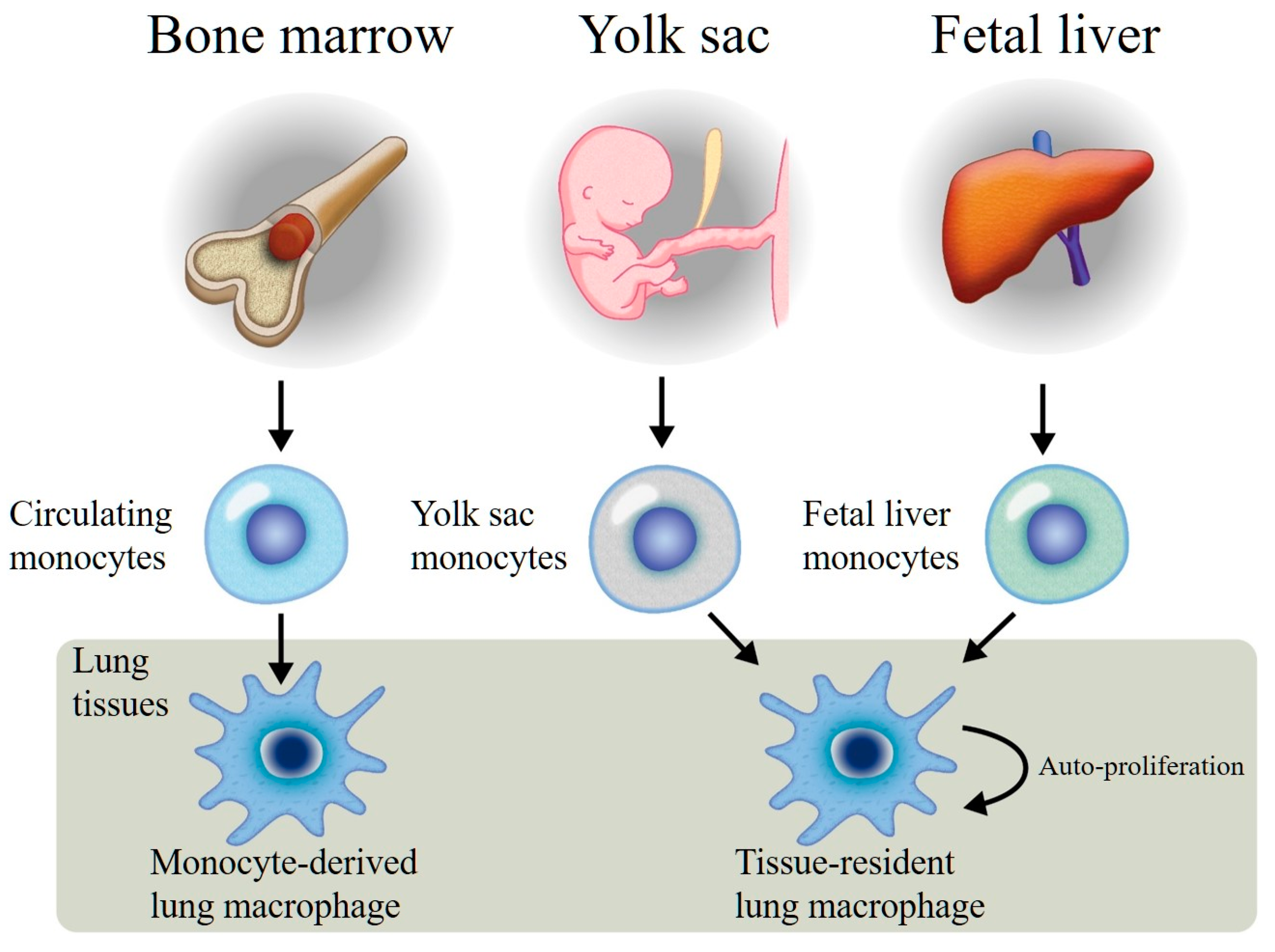
Figure 1.
Monocyte-derived lung macrophages originating from bone marrow pluripotential stem cells, released into the circulating blood, and recruited into the lung tissues. These macrophages have limited proliferation functions. Tissue-resident lung macrophages are generated from both the yolk sac and fetal liver, recruited into the lungs during the early stage of lung development, and become resident lung macrophages with auto-proliferation functions.
Figure 1.
Monocyte-derived lung macrophages originating from bone marrow pluripotential stem cells, released into the circulating blood, and recruited into the lung tissues. These macrophages have limited proliferation functions. Tissue-resident lung macrophages are generated from both the yolk sac and fetal liver, recruited into the lungs during the early stage of lung development, and become resident lung macrophages with auto-proliferation functions.

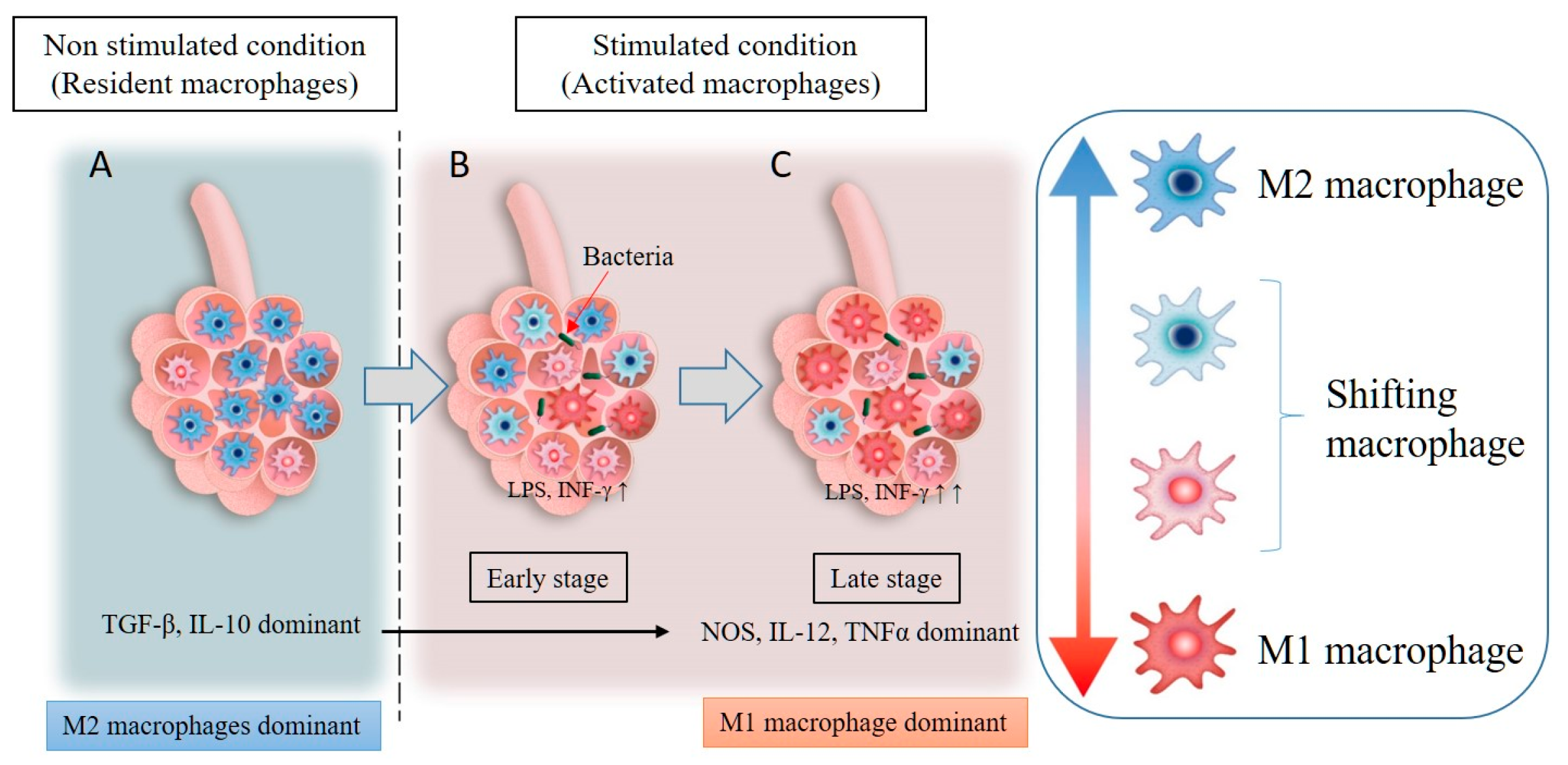
Figure 2.
Resident macrophages are mainly of the M2 phenotype (M2 dominant) under non-stimulated conditions and secrete anti-inflammatory cytokines (TGF-β, IL-10) (A). An inflammatory stimulus in the lungs, such as exposure to pathogens, LPS, particulate matter, or INF-γ, leads to the phenotype switching of resident macrophages from M2 to M1 (B). In addition, circulating monocytes will be recruited into the lung tissues and activated by the inflammatory stimulus, with the majority of the macrophages in the inflammatory zone being M1 macrophages (M1 dominant). These M1 macrophages secrete inflammatory cytokines, such as NOS, IL-12, and TNF-α (C).
Figure 2.
Resident macrophages are mainly of the M2 phenotype (M2 dominant) under non-stimulated conditions and secrete anti-inflammatory cytokines (TGF-β, IL-10) (A). An inflammatory stimulus in the lungs, such as exposure to pathogens, LPS, particulate matter, or INF-γ, leads to the phenotype switching of resident macrophages from M2 to M1 (B). In addition, circulating monocytes will be recruited into the lung tissues and activated by the inflammatory stimulus, with the majority of the macrophages in the inflammatory zone being M1 macrophages (M1 dominant). These M1 macrophages secrete inflammatory cytokines, such as NOS, IL-12, and TNF-α (C).

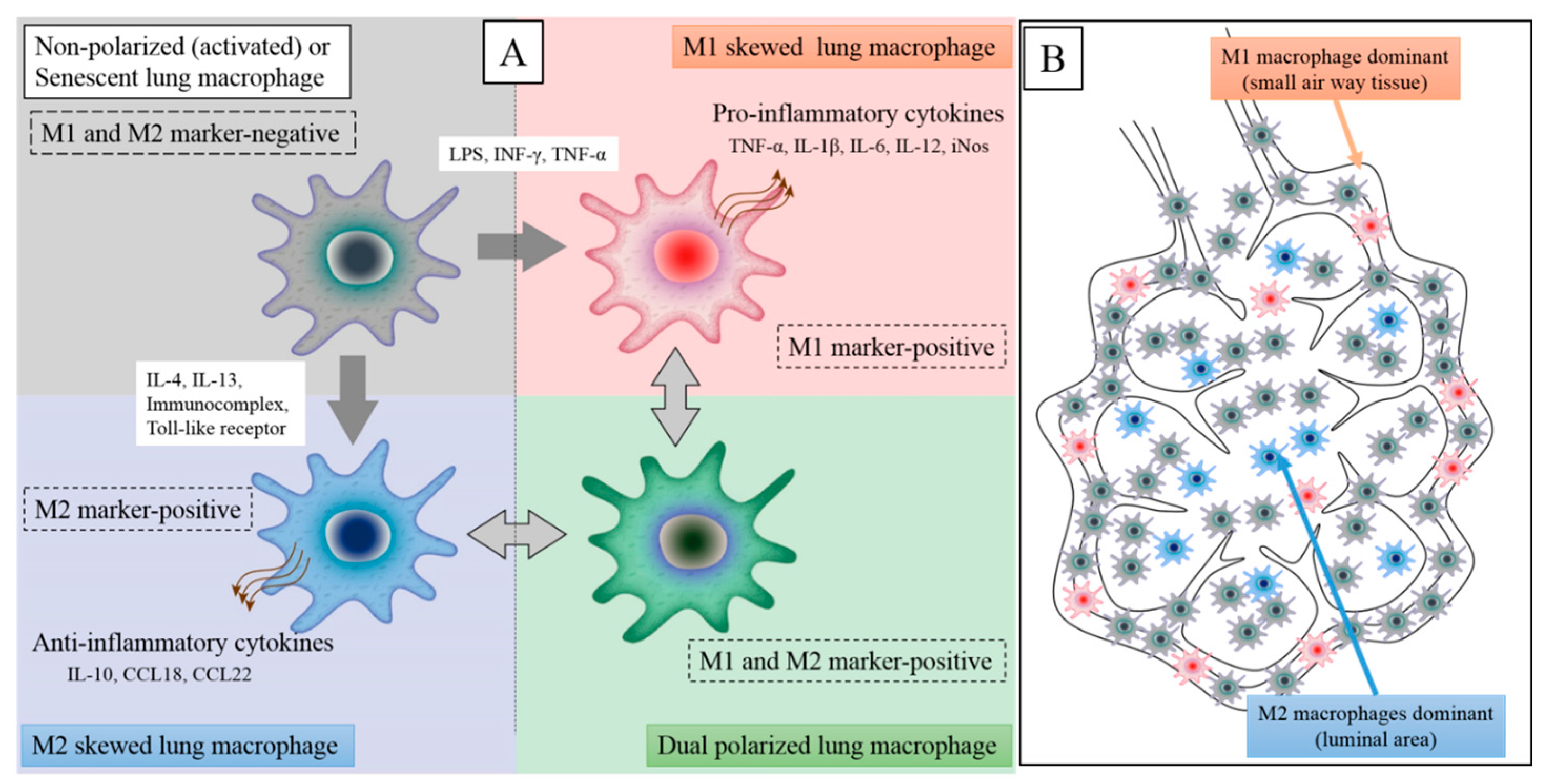
Figure 3.
Patients with COPD have at least four types of macrophages in their lung tissues. Non-polarized or senescent macrophages are M1 and M2 marker-negative and secrete low levels of inflammatory and anti-inflammatory cytokines (left upper). Upon stimulation by Th1 cytokines (e.g., LPS and/or INF-γ), these cell become polarized to predominantly M1 lung macrophages and secrete high levels of pro-inflammatory cytokines (right upper). However, when non-polarized macrophages are stimulated by Th2 cytokines (e.g., IL-4 and/or IL-13), they become polarized to predominantly M2 lung macrophages and secrete high levels of level anti-inflammatory cytokines (left lower). COPD lung tissues also contain a large population of dual-positive macrophages (M1 and M2 marker-positive) (right lower). Nonetheless, the functional properties of dual-positive macrophages are still unclear (A). In COPD lung tissues, M1 macrophages are predominantly found in small airway wall tissues and M2 macrophages in airspaces while abundant non-polarized macrophages are present in both compartments. The distribution of dual-positive macrophages is still unclear (B).
Figure 3.
Patients with COPD have at least four types of macrophages in their lung tissues. Non-polarized or senescent macrophages are M1 and M2 marker-negative and secrete low levels of inflammatory and anti-inflammatory cytokines (left upper). Upon stimulation by Th1 cytokines (e.g., LPS and/or INF-γ), these cell become polarized to predominantly M1 lung macrophages and secrete high levels of pro-inflammatory cytokines (right upper). However, when non-polarized macrophages are stimulated by Th2 cytokines (e.g., IL-4 and/or IL-13), they become polarized to predominantly M2 lung macrophages and secrete high levels of level anti-inflammatory cytokines (left lower). COPD lung tissues also contain a large population of dual-positive macrophages (M1 and M2 marker-positive) (right lower). Nonetheless, the functional properties of dual-positive macrophages are still unclear (A). In COPD lung tissues, M1 macrophages are predominantly found in small airway wall tissues and M2 macrophages in airspaces while abundant non-polarized macrophages are present in both compartments. The distribution of dual-positive macrophages is still unclear (B).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamasaki, K.; Eeden, S.F.v. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. Int. J. Mol. Sci. 2018, 19, 582. https://doi.org/10.3390/ijms19020582
AMA Style
Yamasaki K, Eeden SFv. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. International Journal of Molecular Sciences. 2018; 19(2):582. https://doi.org/10.3390/ijms19020582
Chicago/Turabian StyleYamasaki, Kei, and Stephan F. van Eeden. 2018. "Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD" International Journal of Molecular Sciences 19, no. 2: 582. https://doi.org/10.3390/ijms19020582
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.