Core Microbiome of Medicinal Plant Salvia miltiorrhiza Seed: A Rich Reservoir of Beneficial Microbes for Secondary Metabolism?

 ,
,
Abstract
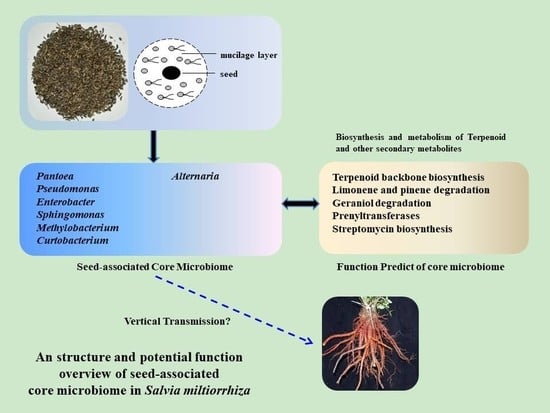
:
1. Introduction
2. Results
2.1. Genetic Diversity of Different S. miltiorrhiza Seeds
2.2. The Bacterial 16S rRNA and Fungal ITS Sequencing Data Set
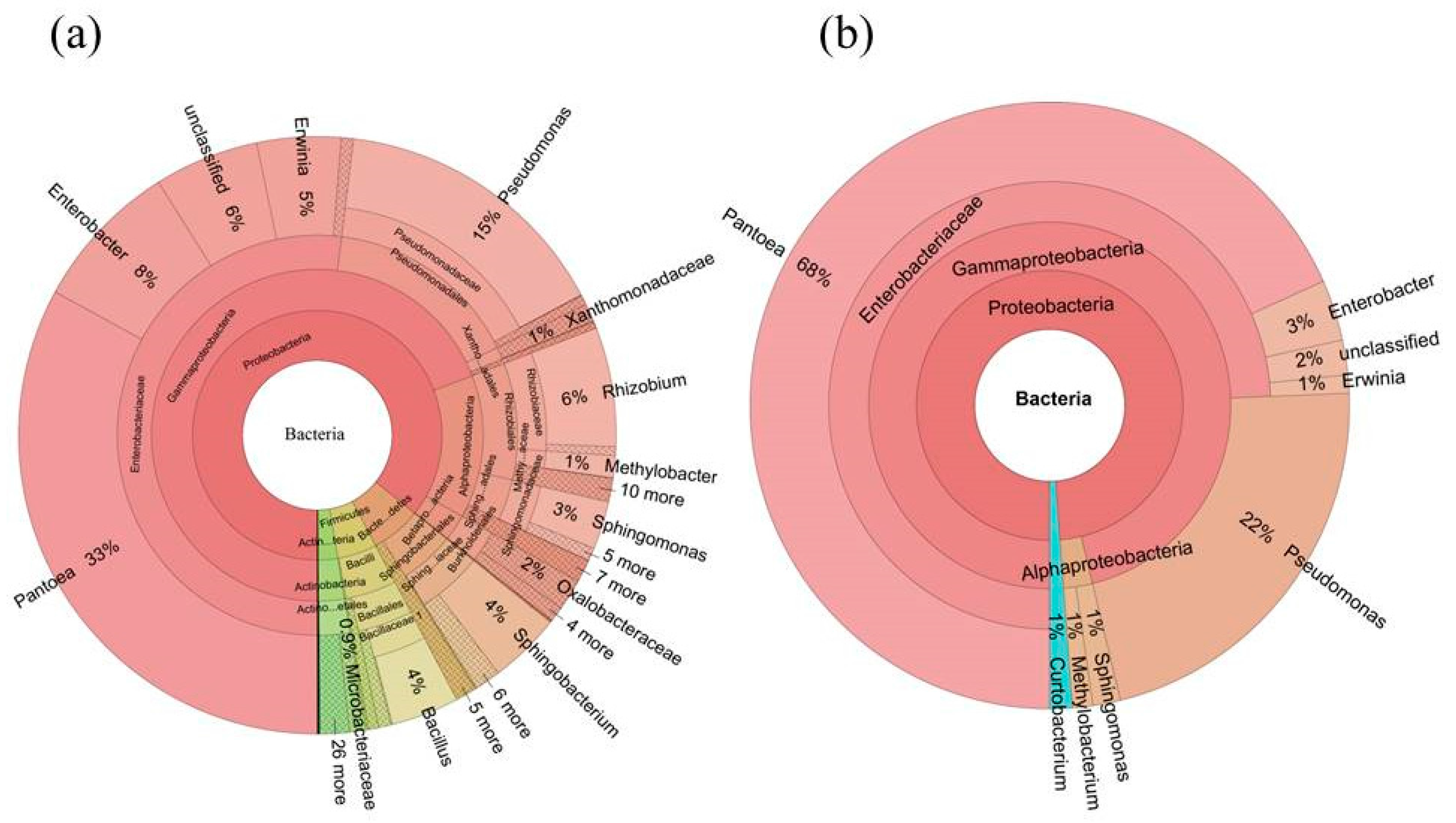
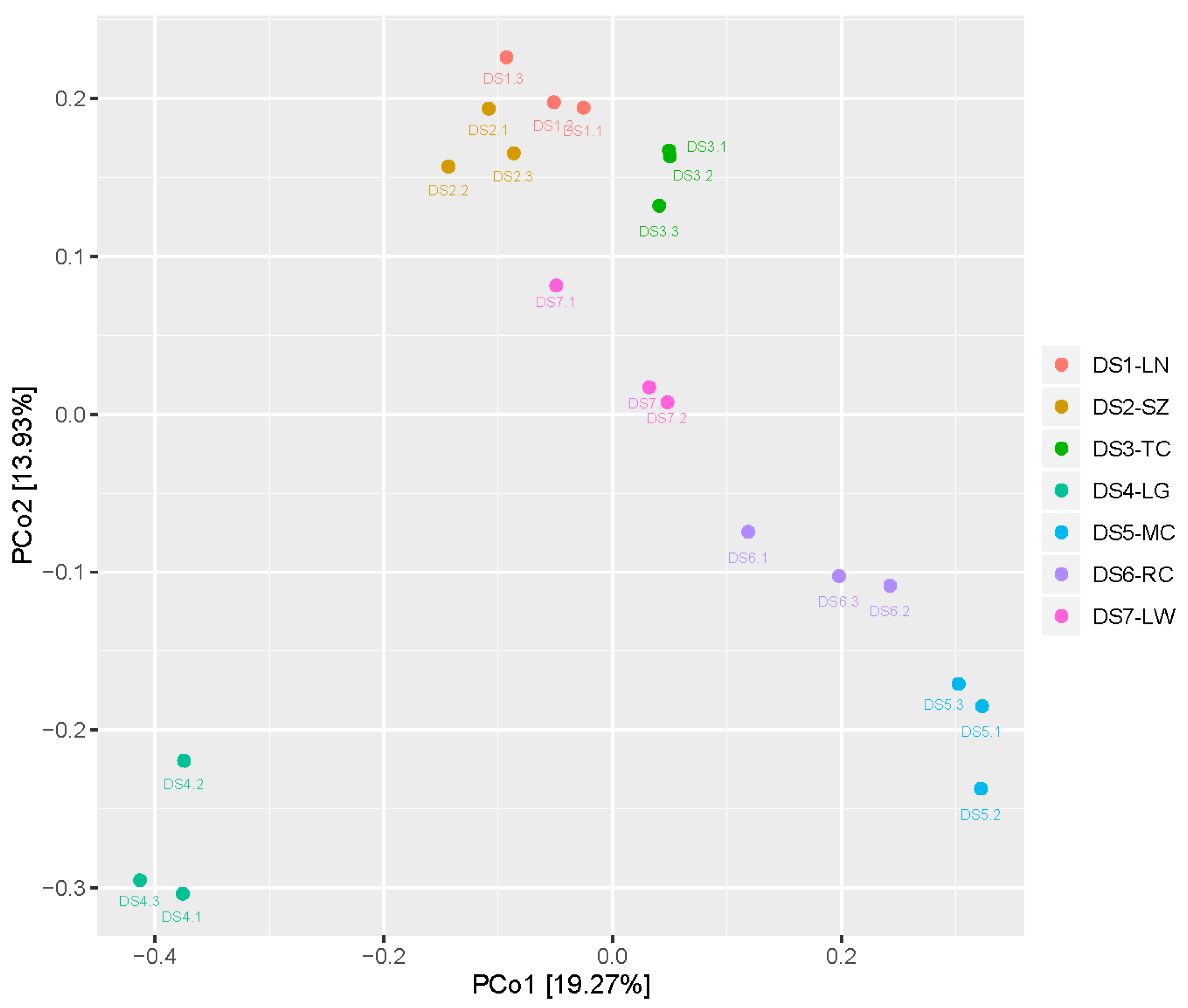
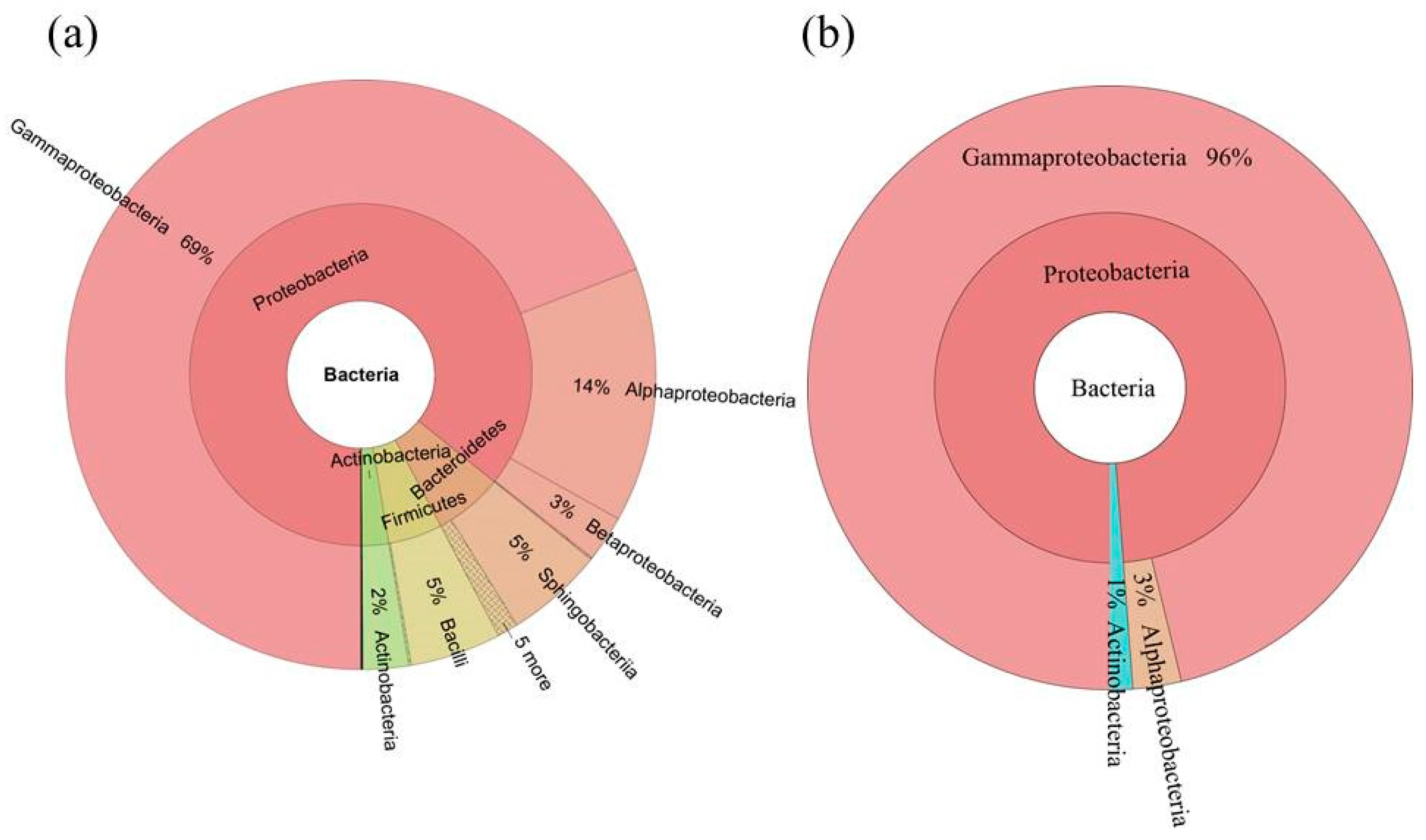
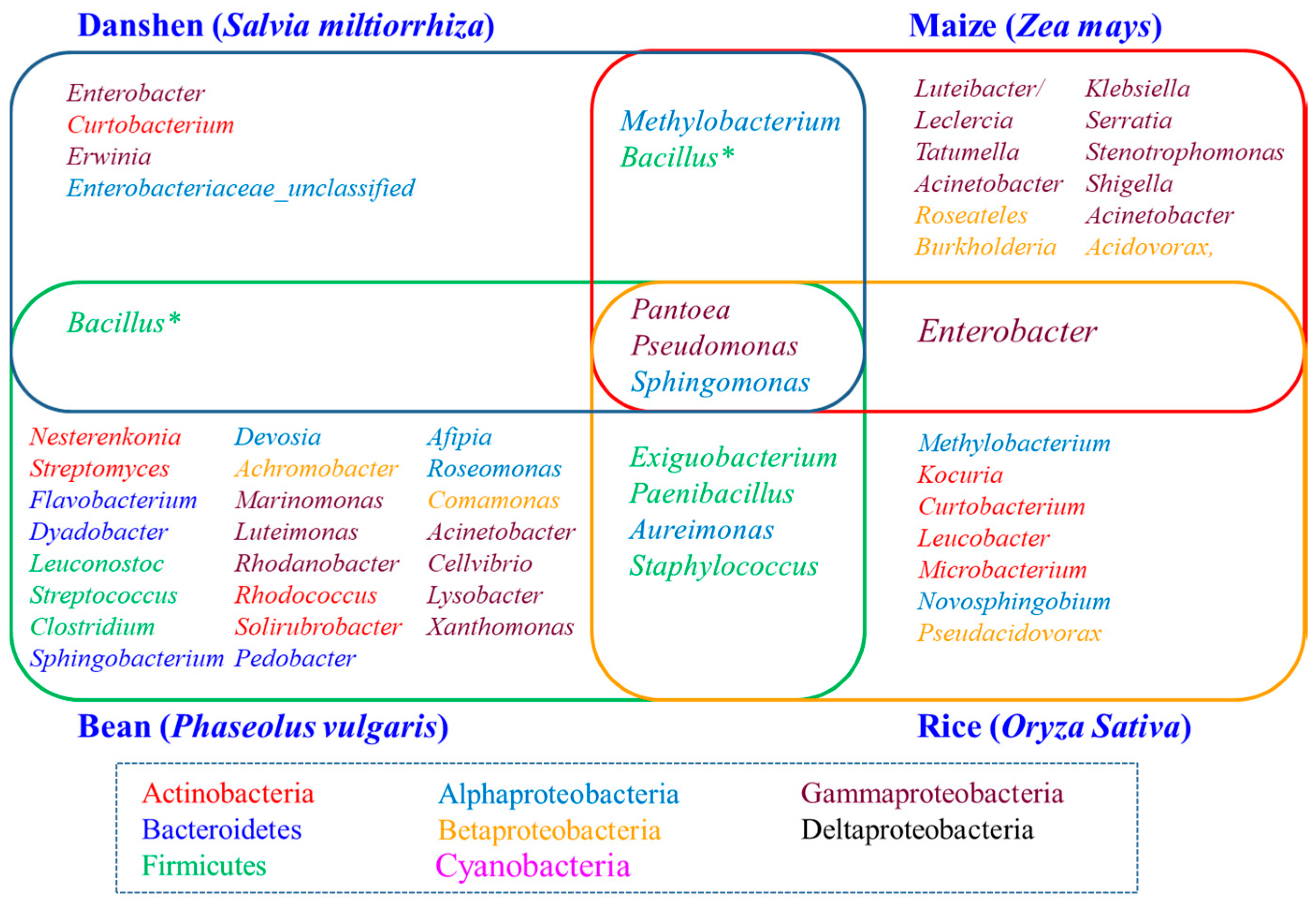
2.3. Diversity of the Seed-Associated Bacterial Microbiome in S. miltiorrhiza
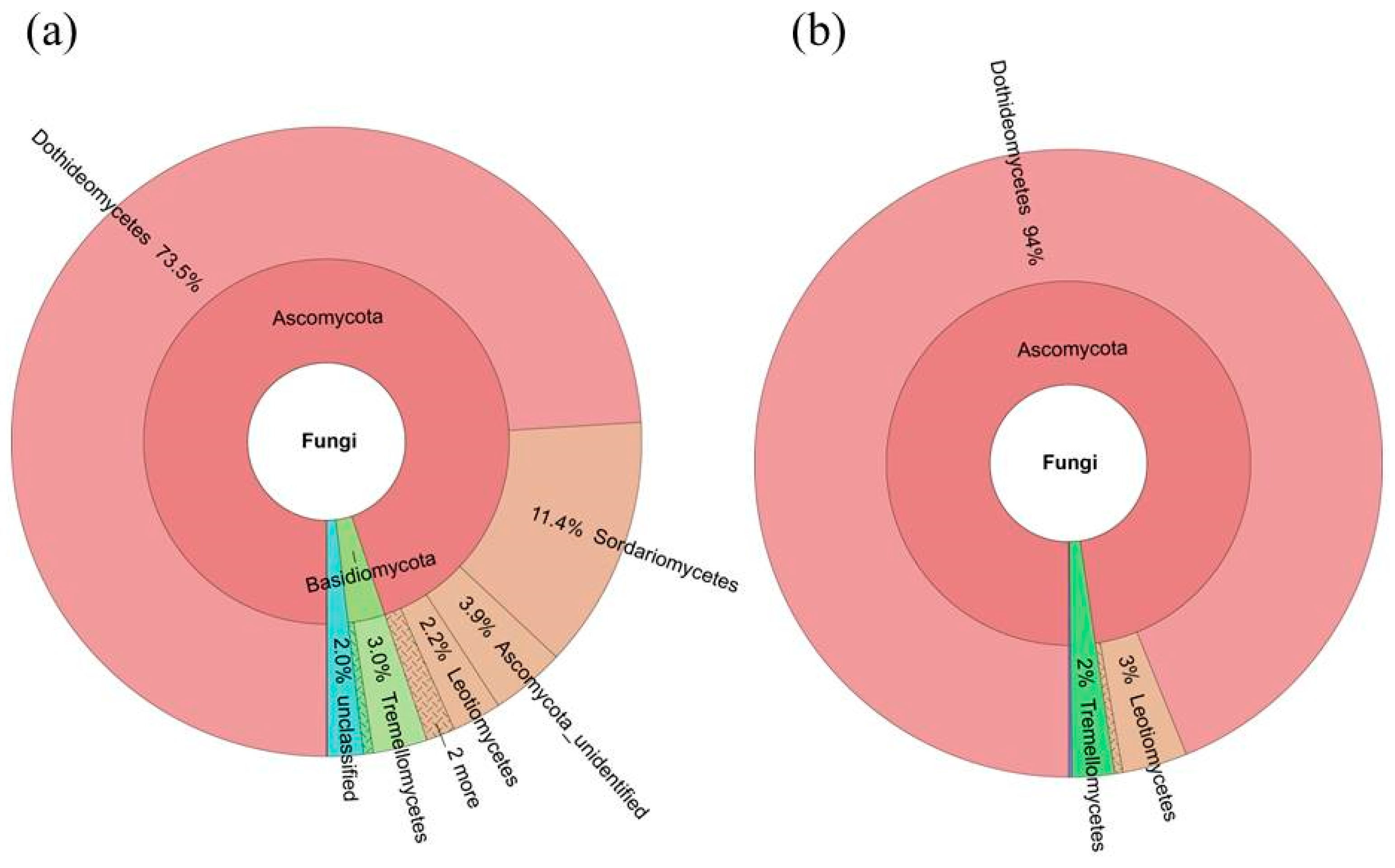
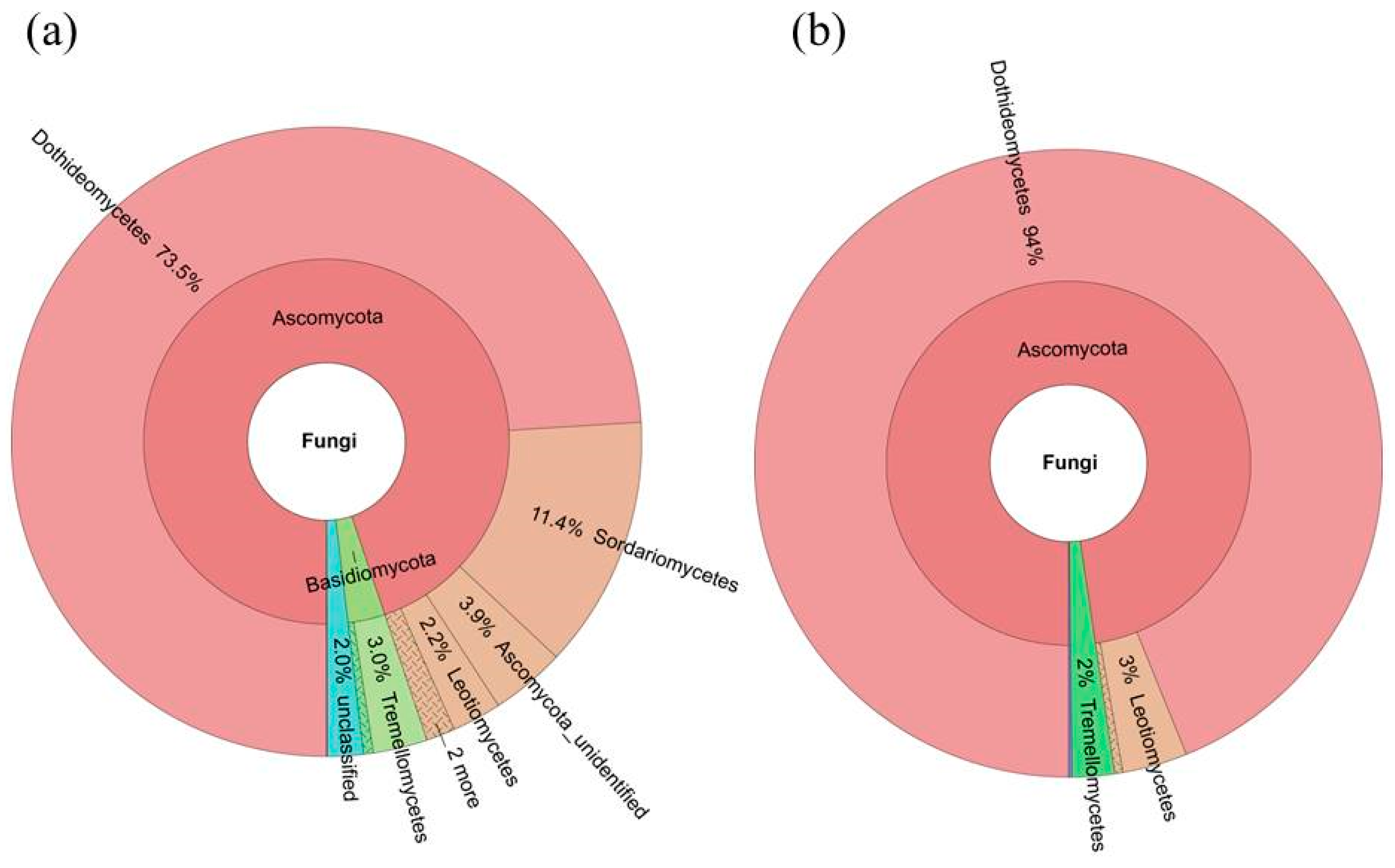
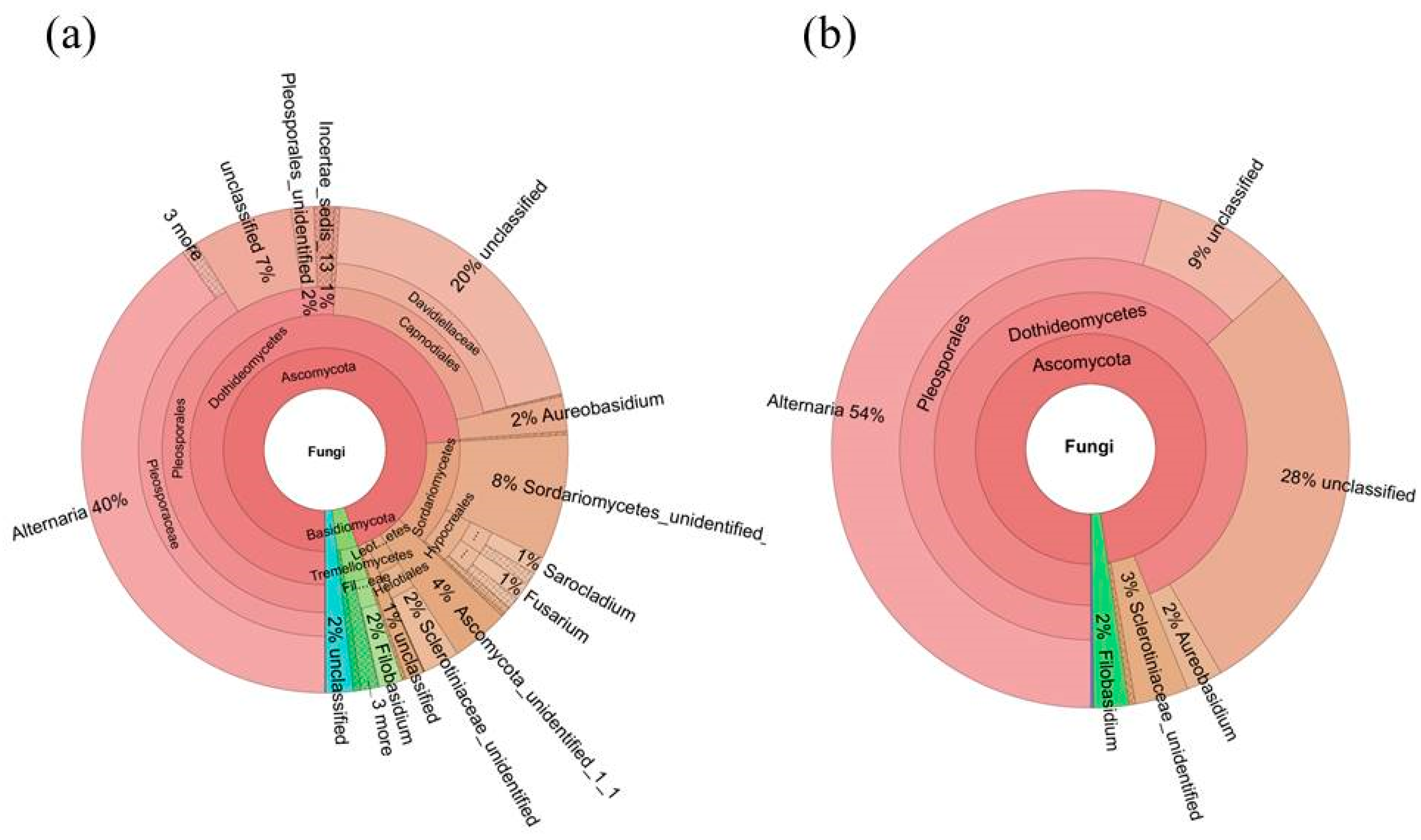
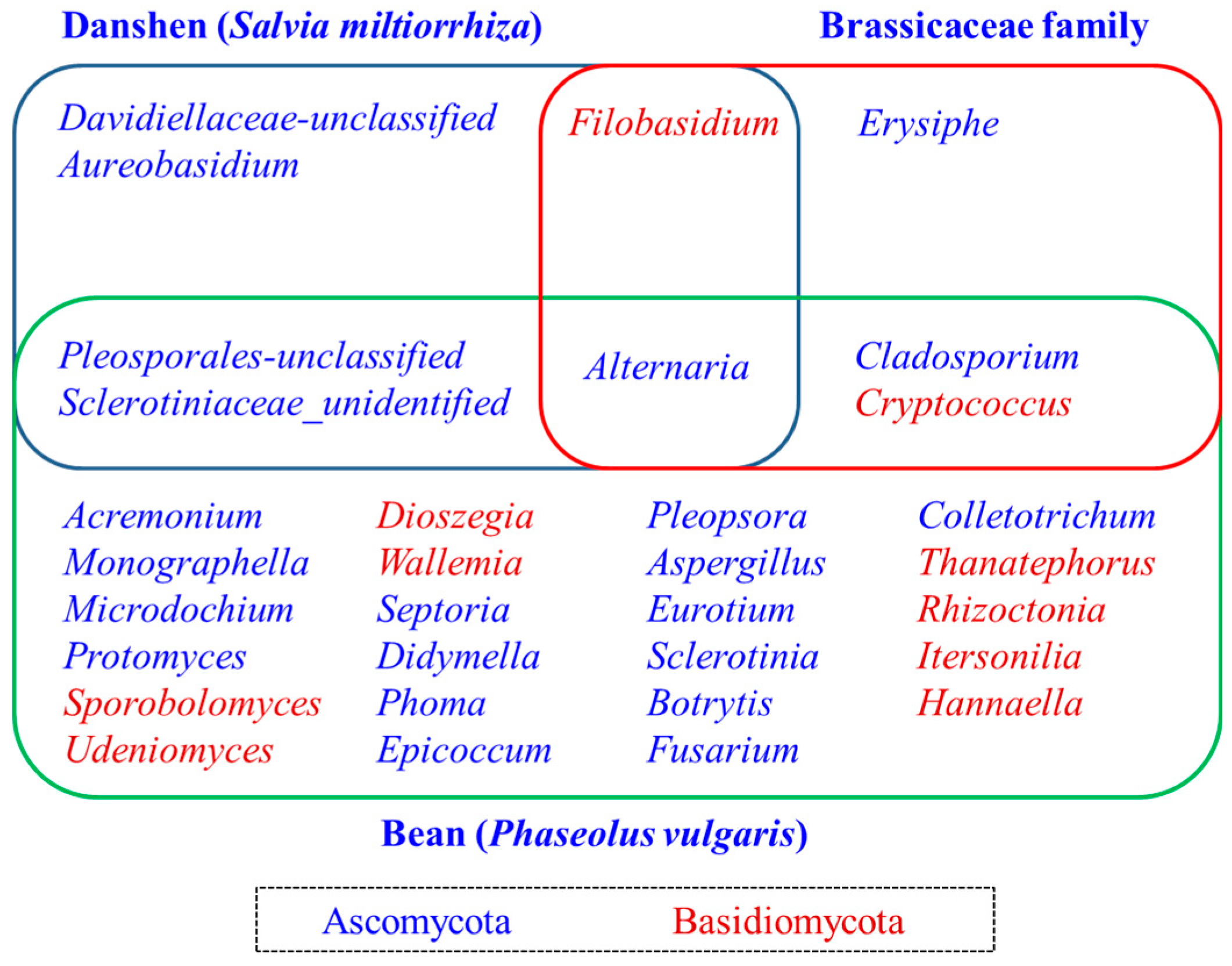
2.4. Diversity of the Seed-Associated Fungal Microbiome in S. miltiorrhiza
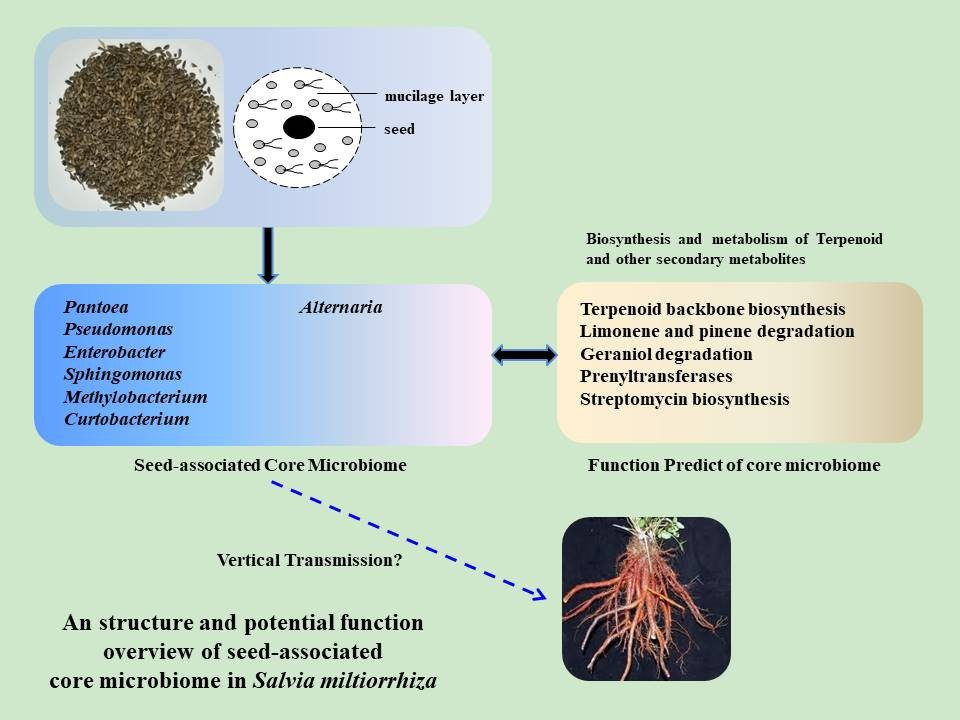
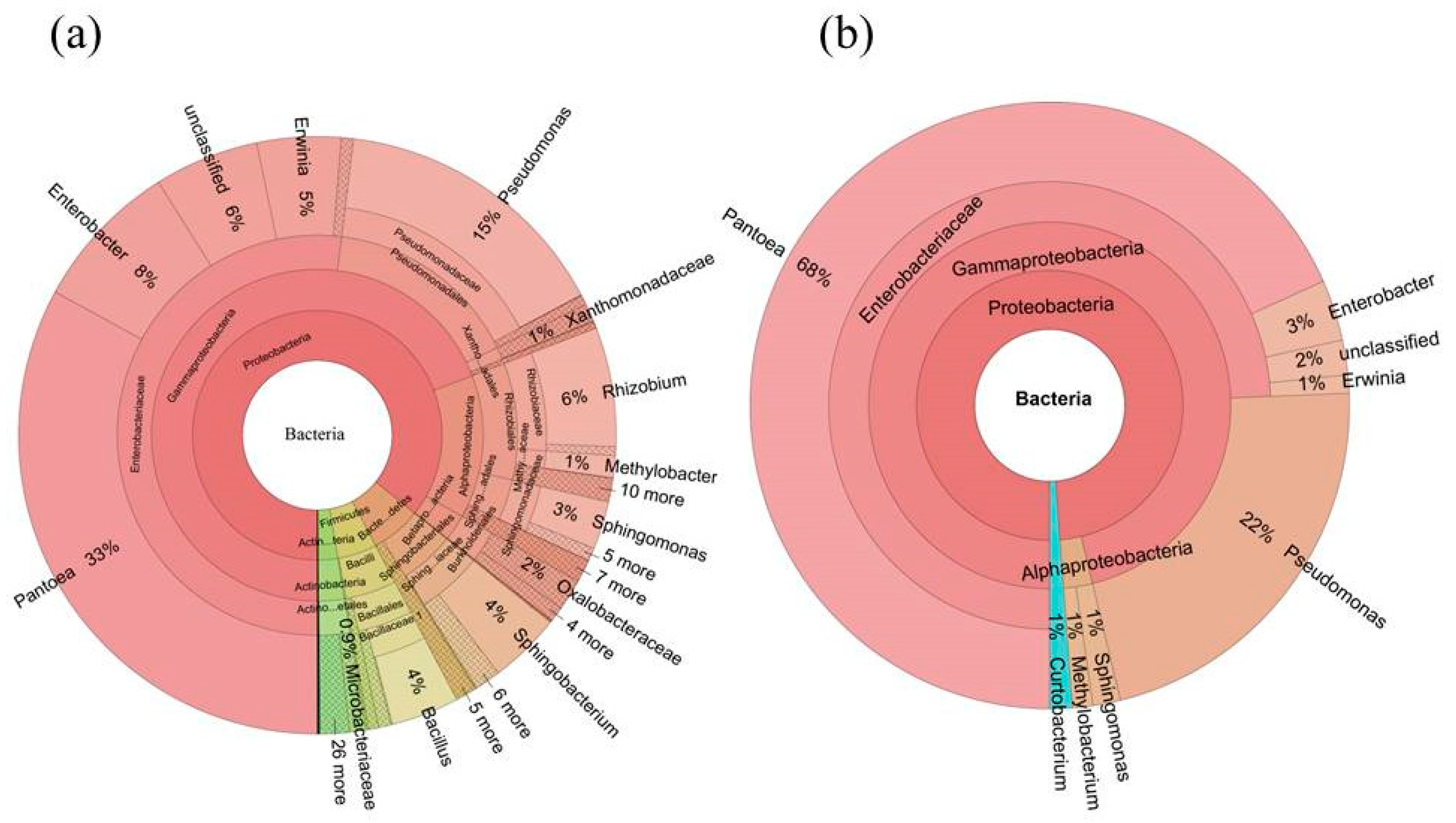
2.5. Determination of the Core Bacterial Microbiome (Bacteriome) of S. miltiorrhiza Seeds
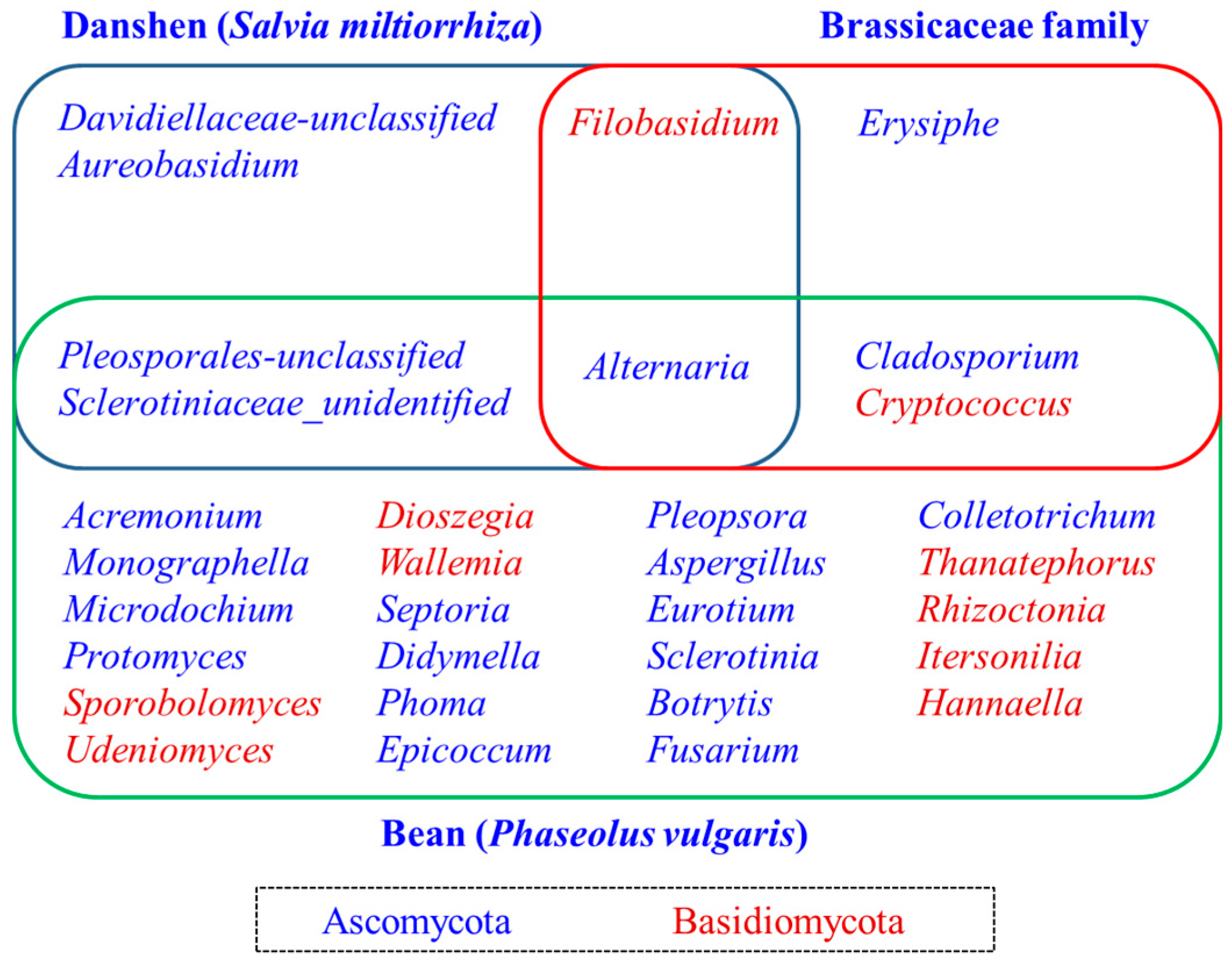
2.6. Determination of the Core Fungal Microbiome (Mycobiome) of S. miltiorrhiza Seeds
2.7. Predictive Function of Core Bacterial Microbiome in S. miltiorrhiza Seeds
3. Discussion
4. Material and Methods
4.1. Sampling of Salvia miltiorrhiza Seeds
4.2. Plant DNA Exaction and SSR Genotyping
4.3. Microbial DNA Extraction
4.4. PCR Amplification, 16S rDNA or ITS Sequencing and Data Analysis
4.5. Determination of Core Microbiome of S. miltiorrhiza Seed
4.6. Predict Microbial Functional Profiles of Core Microbiome
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nelson, E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil 2017, 422, 7–34. [Google Scholar] [CrossRef]
- Shade, A.; Jacques, M.A.; Barret, M. Ecological patterns of seed microbiome diversity, transmission, and assembly. Curr. Opin. Microbiol. 2017, 37, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Truyens, S.; Weyens, N.; Cuypers, A.; Vangronsveld, J. Bacterial seed endophytes: Genera, vertical transmission and interaction with plants. Environ. Microbiol. Rep. 2015, 7, 40–50. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Astudillo-Garcia, C.; Bell, J.J.; Webster, N.S.; Glasl, B.; Jompa, J.; Montoya, J.M.; Taylor, M.W. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 2017, 19, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Garrido-Oter, R.; Munch, P.C.; Weiman, A.; Droge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911-20. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Backhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Kratz, M.; Damman, C.J.; Hullarg, M. Mechanisms linking the gut microbiome and glucose metabolism. J. Clin. Endocrinol. Metab. 2016, 101, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Koonen, D.; Hofker, M.; Fu, J. Gut microbiome and lipid metabolism: From associations to mechanisms. Curr. Opin. Lipidol. 2016, 27, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Klaedtke, S.; Jacques, M.A.; Raggi, L.; Preveaux, A.; Bonneau, S.; Negri, V.; Chable, V.; Barret, M. Terroir is a key driver of seed-associated microbial assemblages. Environ. Microbiol. 2016, 18, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Barret, M.; Briand, M.; Bonneau, S.; Preveaux, A.; Valiere, S.; Bouchez, O.; Hunault, G.; Simoneau, P.; Jacquesa, M.A. Emergence shapes the structure of the seed microbiota. Appl. Environ. Microbiol. 2015, 81, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Links, M.G.; Demeke, T.; Grafenhan, T.; Hill, J.E.; Hemmingsen, S.M.; Dumonceaux, T.J. Simultaneous profiling of seed-associated bacteria and fungi reveals antagonistic interactions between microorganisms within a shared epiphytic microbiome on Triticum and Brassica seeds. New Phytol. 2014, 202, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, E.M.; Raizada, M.N. Taxonomic and functional diversity of cultured seed associated microbes of the cucurbit family. BMC Microbiol. 2016, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Yeh, M.W.; Hsieh, Y.T.; Chung, W.C.; Lo, C.T.; Young, L.S. Diversity and functional characterization of bacterial endophytes dwelling in various rice (Oryza sativa L.) tissues, and their seed-borne dissemination into rhizosphere under gnotobiotic P-stress. Plant Soil 2015, 394, 177–197. [Google Scholar] [CrossRef]
- Sorty, A.M.; Meena, K.K.; Choudhary, K.; Bitla, U.M.; Minhas, P.S.; Krishnani, K.K. Effect of plant growth promoting bacteria associated with halophytic weed (Psoralea corylifolia L.) on germination and seedling growth of wheat under saline conditions. Appl. Biochem. Biotechnol. 2016, 180, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Herrera, S.D.; Grossi, C.; Zawoznik, M.; Groppa, M.D. Wheat seeds harbour bacterial endophytes with potential as plant growth promoters and biocontrol agents of Fusarium graminearum. Microbiol. Res. 2016, 186, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Chimwamurombe, P.M.; Gronemeyer, J.L.; Reinhold-Hurek, B. Isolation and characterization of culturable seed-associated bacterial endophytes from gnotobiotically grown Marama bean seedlings. FEMS Microbiol. Ecol. 2016, 92, fiw083. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, A.S.; Pintelon, I.; Stevens, V.; Imperato, V.; Timmermans, J.P.; Gonzalez-Chavez, C.; Carrillo-Gonzalez, R.; van Hamme, J.; Vangronsveld, J.; Thijs, S. Seed endophyte microbiome of Crotalaria pumila unpeeled: Identification of plant-beneficial methylobacteria. Int. J. Mol. Sci. 2018, 19, 291. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.J.J.; Caradus, J.R.; Johnson, L.J. Fungal endophytes for sustainable crop production. FEMS Microbiol. Ecol. 2016, 92, fiw194. [Google Scholar] [CrossRef] [PubMed]
- Su, C.Y.; Ming, Q.L.; Rahman, K.; Han, T.; Qin, L.P. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar] [CrossRef]
- Li, X.Q.; Zhai, X.; Shu, Z.H.; Dong, R.F.; Ming, Q.L.; Qin, L.P.; Zheng, C.J. Phoma glomerata D14: An endophytic fungus from Salvia miltiorrhiza that produces salvianolic acid C. Curr. Microbiol. 2016, 73, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ming, Q.; Han, T.; Li, W.; Zhang, Q.; Zhang, H.; Zheng, C.; Huang, F.; Rahman, K.; Qin, L. Tanshinone IIA and tanshinone I production by Trichoderma atroviride D16, an endophytic fungus in Salvia miltiorrhiza. Phytomedicine 2012, 19, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, S.C.; Zhang, J.Y.; Ma, P.D.; Duan, J.L.; Liang, Z.S. Effect and mechanism of endophytic bacteria on growth and secondary metabolite synthesis in Salvia miltiorrhiza hairy roots. Acta Physiol. Plant. 2014, 36, 1095–1105. [Google Scholar] [CrossRef]
- Sun, J.; Xia, F.; Cui, L.; Liang, J.; Wang, Z.; Wei, Y. Characteristics of foliar fungal endophyte assemblages and host effective components in Salvia miltiorrhiza Bunge. Appl. Microbiol. Biotechnol. 2014, 98, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; He, L.; Song, G.; Wang, R. Antagonistic bioactivity of endophytic strains isolated from Salvia miltiorrhiza. Afr. J. Biotechnol. 2013, 10, 15117–15122. [Google Scholar] [CrossRef]
- Ming, Q.; Su, C.; Zheng, C.; Jia, M.; Zhang, Q.; Zhang, H.; Rahman, K.; Han, T.; Qin, L. Elicitors from the endophytic fungus Trichoderma atroviride promote Salvia miltiorrhiza hairy root growth and tanshinone biosynthesis. J. Exp. Bot. 2013, 64, 5687–5694. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Tang, H.Y.; Duan, J.L.; Gao, J.M.; Xue, Q.H. Bioactive alkaloids produced by Pseudomonas brassicacearum subsp. Neoaurantiaca, an endophytic bacterium from Salvia miltiorrhiza. Nat. Prod. Res. 2013, 27, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Yuan, H.M.; Qi, S.S.; Feng, M.L.; Liu, H.W.; Zhang, X.P. Isolation and genetic diversity of the endophytic actinomycetes from Salvia miltiorrhiza Bge. and Polygonatum sibiricum Red. Microbiol. China 2010, 37, 1341–1346. [Google Scholar]
- He, X.L.; Wang, L.Y.; Ma, J.; Zhao, L.L. AM fungal diversity in the rhizosphere of Salvia miltiorrhiza in Anguo city of Hebei province. Biodivers. Sci. 2010, 18, 187–194. [Google Scholar]
- Liu, Y.; Zuo, S.; Zou, Y.Y.; Wang, J.H.; Song, W. Investigation on diversity and population succession dynamics of endophytic bacteria from seeds of maize (Zea mays L. Nongda108) at different growth stages. Ann. Microbiol. 2013, 63, 71–79. [Google Scholar] [CrossRef]
- Midha, S.; Bansal, K.; Sharma, S.; Kumar, N.; Patil, P.P.; Chaudhry, V.; Patil, P.B. Genomic resource of rice seed associated bacteria. Front. Microbiol. 2016, 6, 1551. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Danzberger, J.; Scholer, A.; Schroder, P.; Schloter, M.; Radl, V. Dominant groups of potentially active bacteria shared by barley seeds become less abundant in root associated microbiome. Front. Plant Sci. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, D.; Mancinelli, R.; Wikström, M.; Birch-Jensen, A.-S.; Postma, J.; Ehlers, R.-U.; Goertz, S.; Berg, G. The structure of the Brassica napus seed microbiome is cultivar-dependent and affects the interactions of symbionts and pathogens. Microbiome 2017, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, R.; Niu, Y.; Lin, H.; Ye, W.; Guo, L.; Hu, X. Whole genome sequence of Pantoea ananatis R100, an antagonistic bacterium isolated from rice seed. J. Biotechnol. 2016, 225, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Sheibani-Tezerji, R.; Naveed, M.; Jehl, M.A.; Sessitsch, A.; Rattei, T.; Mitter, B. The genomes of closely related Pantoea ananatis maize seed endophytes having different effects on the host plant differ in secretion system genes and mobile genetic elements. Front. Microbiol. 2015, 6, 440. [Google Scholar] [CrossRef] [PubMed]
- Town, J.; Dumonceaux, T.J. High-quality draft genome sequences of Pantoea agglomerans isolates exhibiting antagonistic interactions with wheat seed-associated fungi. Genome Announc. 2016, 4, e00511–e00516. [Google Scholar] [CrossRef] [PubMed]
- Zamioudis, C.; Mastranesti, P.; Dhonukshe, P.; Blilou, I.; Pieterse, C.M. Unraveling root developmental programs initiated by beneficial Pseudomonas spp. bacteria. Plant Physiol. 2013, 162, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Van de Mortel, J.E.; de Vos, R.C.; Dekkers, E.; Pineda, A.; Guillod, L.; Bouwmeester, K.; van Loon, J.J.; Dicke, M.; Raaijmakers, J.M. Metabolic and transcriptomic changes induced in arabidopsis by the rhizobacterium Pseudomonas fluorescens SS101. Plant Physiol. 2012, 160, 2173–2188. [Google Scholar] [CrossRef] [PubMed]
- Sessitsch, A.; Hardoim, P.; Doring, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M.; et al. Functional Characteristics of an Endophyte Community Colonizing Rice Roots as Revealed by Metagenomic Analysis. Mol. Plant Microbe 2012, 25, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Hartman, K.; van der Heijden, M.G.; Roussely-Provent, V.; Walser, J.C.; Schlaeppi, K. Deciphering composition and function of the root microbiome of a legume plant. Microbiome 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Meng, Q.; Wang, Q.; Luo, S.; Chen, B.; Khan, K.Y.; Yang, X.; Feng, Y. Endophytic bacterium Sphingomonas SaMR12 promotes cadmium accumulation by increasing glutathione biosynthesis in Sedum alfredii Hance. Chemosphere 2016, 154, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.L.; Sun, P.; Wang, X.M.; Cheng, S.; Lv, F.; Qiu, T.L.; Yuan, M.; Sun, J.G. Sphingomonaszeicaulis sp. nov. an endophytic bacterium isolated from maize root. Int. J. Syst. Evol. Microbiol. 2016, 66, 3755–3760. [Google Scholar] [PubMed]
- Ismaiel, A.A.; Ahmed, A.S.; Hassan, I.A.; El-Sayed, E.R.; Karam El-Din, A.A. Production of paclitaxel with anticancer activity by two local fungal endophytes, Aspergillus fumigatus and Alternaria tenuissima. Appl. Microbiol. Biotechnol. 2017, 101, 5831–5846. [Google Scholar] [CrossRef] [PubMed]
- Bian, G.; Yuan, Y.; Tao, H.; Shi, X.; Zhong, X.; Han, Y.; Fu, S.; Fang, C.; Deng, Z.; Liu, T. Production of taxadiene by engineering of mevalonate pathway in Escherichia coli and endophytic fungus Alternaria alternata TPF6. Biotechnol. J. 2017, 12, 1600697. [Google Scholar] [CrossRef] [PubMed]
- Soltani, J.; Hosseyni Moghaddam, M.S. Antiproliferative, antifungal, and antibacterial activities of endophytic Alternaria species from cupressaceae. Curr. Microbiol. 2014, 69, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.M.; Kaur, A.; Raja, H.A.; Kellogg, J.J.; Oberlies, N.H.; Cech, N.B. Antimicrobial fungal endophytes from the botanical medicine goldenseal (Hydrastis canadensis). Phytochem. Lett. 2016, 17, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Winkelblech, J.; Fan, A.; Li, S.M. Prenyltransferases as key enzymes in primary and secondary metabolism. Appl. Microbiol. Biotechnol. 2015, 99, 7379–7397. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, L.; Massardo, D.R.; Pontieri, P.; Bertea, C.M.; Mombello, D.; Carata, E.; Tredici, S.M.; Talà, A.; Mucciarelli, M.; Groudeva, V.I.; et al. The microbial community of Vetiver root and its involvement into essential oil biogenesis. Environ. Microbiol. 2008, 10, 2824–2841. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Shen, C.; Pan, Y.Y.; Han, Y.W.; Yang, M.; Yu, W.D.; Liu, J.L.; Wu, X.; Li, M.D.; Qi, Z.C.; et al. Development of 40 novel microsatellites in wild populations of Salvia miltiorrhiza Burge based on a transcriptome database. Conserv. Genet. Resour. 2017. accepted. [Google Scholar]
- Qi, Z.C.; Shen, C.; Han, Y.W.; Shen, W.; Yang, M.; Liu, J.; Liang, Z.S.; Li, P.; Fu, C.X. Development of microsatellite loci in Mediterranean sarsaparilla (Smilax aspera; Smilacaceae) using transcriptome data. Appl. Plant Sci. 2017, 5, 1700005. [Google Scholar] [CrossRef] [PubMed]
- Narzary, D.; Verma, S.; Mahar, K.S.; Rana, T.S. A rapid and effective method for isolation of genomic DNA from small amount of silica-dried leaf tissues. Natl. Acad. Sci. Lett. 2015, 38, 441–444. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina paired-end read merger. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.; Gu, Y.Q.; Coleman-Derr, D. MetaCoMET: A web platform for discovery and visualization of the core microbiome. Bioinformatics 2016, 32, 3469–3470. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Aleti, G.; Nikolić, B.; Brader, G.; Pandey, R.V.; Antonielli, L.; Pfeiffer, S.; Oswald, A.; Sessitsch, A. Secondary metabolite genes encoded by potato rhizosphere microbiomes in the Andean highlands are diverse and vary with sampling site and vegetation stage. Sci. Rep. 2017, 7, 2330. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Wu, H.; Yan, B.; Zhao, H.; Liu, F.; Zhang, H.; Sheng, Q.; Miao, F.; Liang, Z. Core Microbiome of Medicinal Plant Salvia miltiorrhiza Seed: A Rich Reservoir of Beneficial Microbes for Secondary Metabolism? Int. J. Mol. Sci. 2018, 19, 672. https://doi.org/10.3390/ijms19030672
Chen H, Wu H, Yan B, Zhao H, Liu F, Zhang H, Sheng Q, Miao F, Liang Z. Core Microbiome of Medicinal Plant Salvia miltiorrhiza Seed: A Rich Reservoir of Beneficial Microbes for Secondary Metabolism? International Journal of Molecular Sciences. 2018; 19(3):672. https://doi.org/10.3390/ijms19030672
Chicago/Turabian StyleChen, Haimin, Hongxia Wu, Bin Yan, Hongguang Zhao, Fenghua Liu, Haihua Zhang, Qing Sheng, Fang Miao, and Zongsuo Liang. 2018. "Core Microbiome of Medicinal Plant Salvia miltiorrhiza Seed: A Rich Reservoir of Beneficial Microbes for Secondary Metabolism?" International Journal of Molecular Sciences 19, no. 3: 672. https://doi.org/10.3390/ijms19030672