Integrated Molecular Characterization of Gastrointestinal Stromal Tumors (GIST) Harboring the Rare D842V Mutation in PDGFRA Gene

 , , , ,
, , , ,  ,
,
Abstract
:
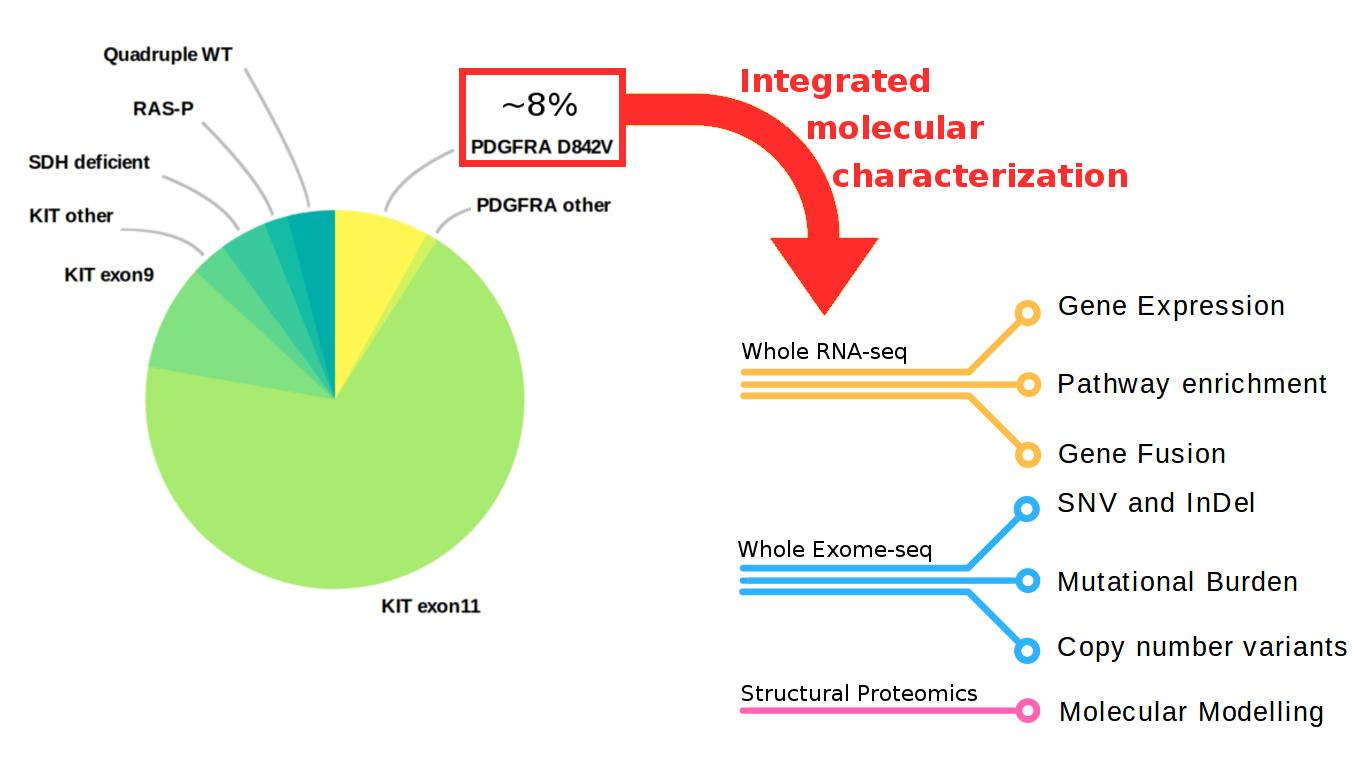
1. Introduction
2. Results
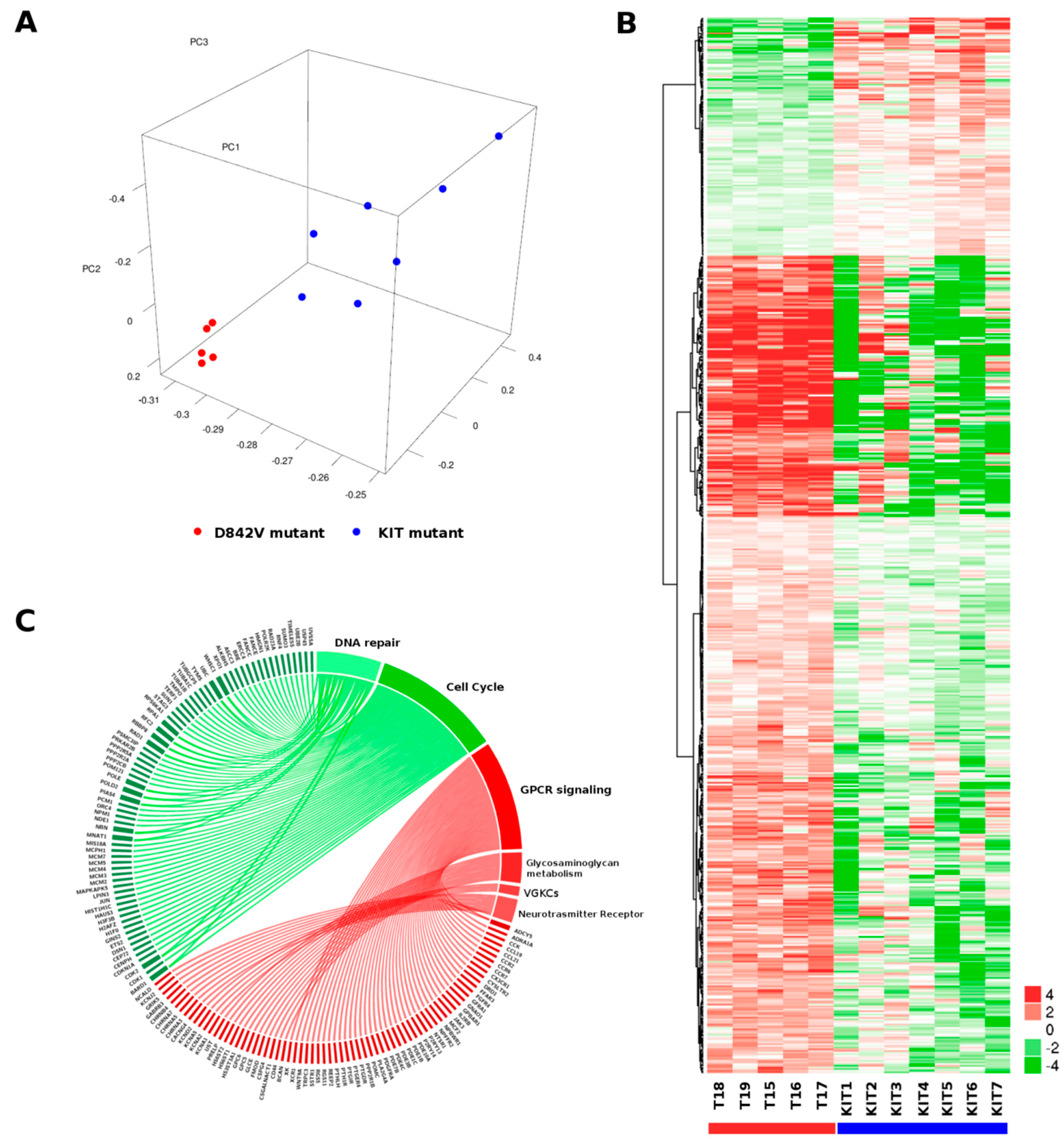
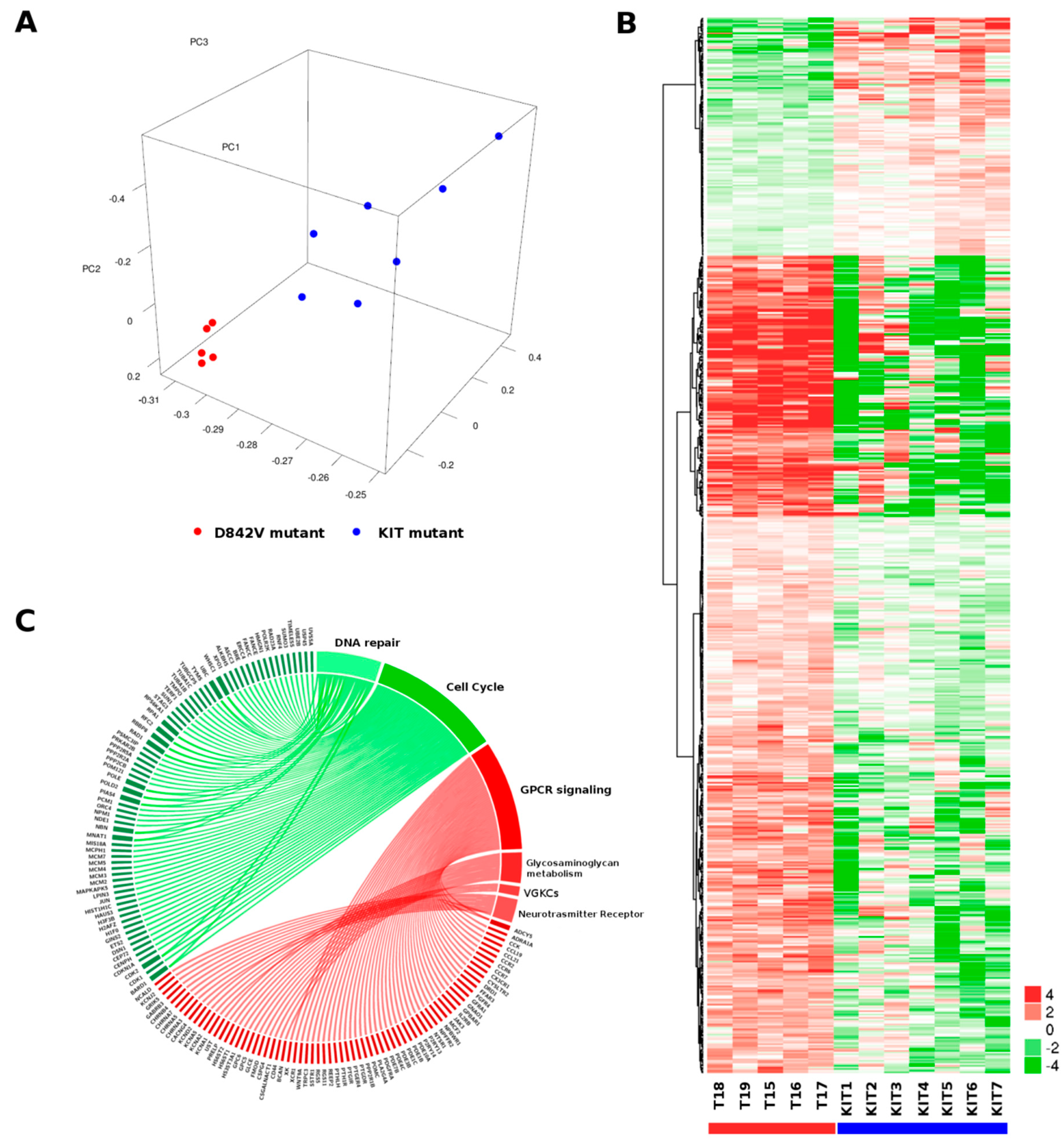
2.1. Gene Expression Profile
2.2. Fusion Transcript by Whole Transcriptome Sequencing
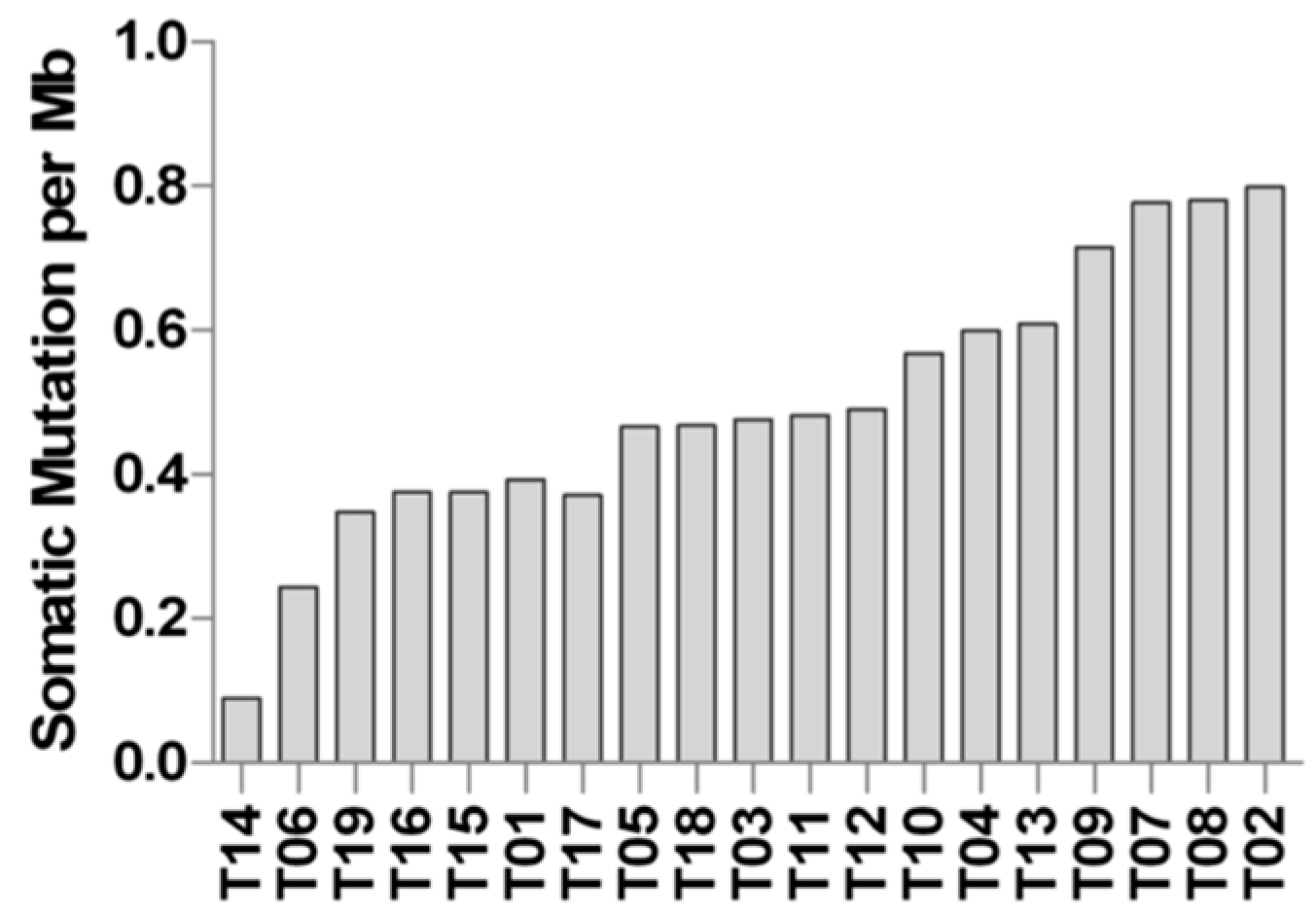
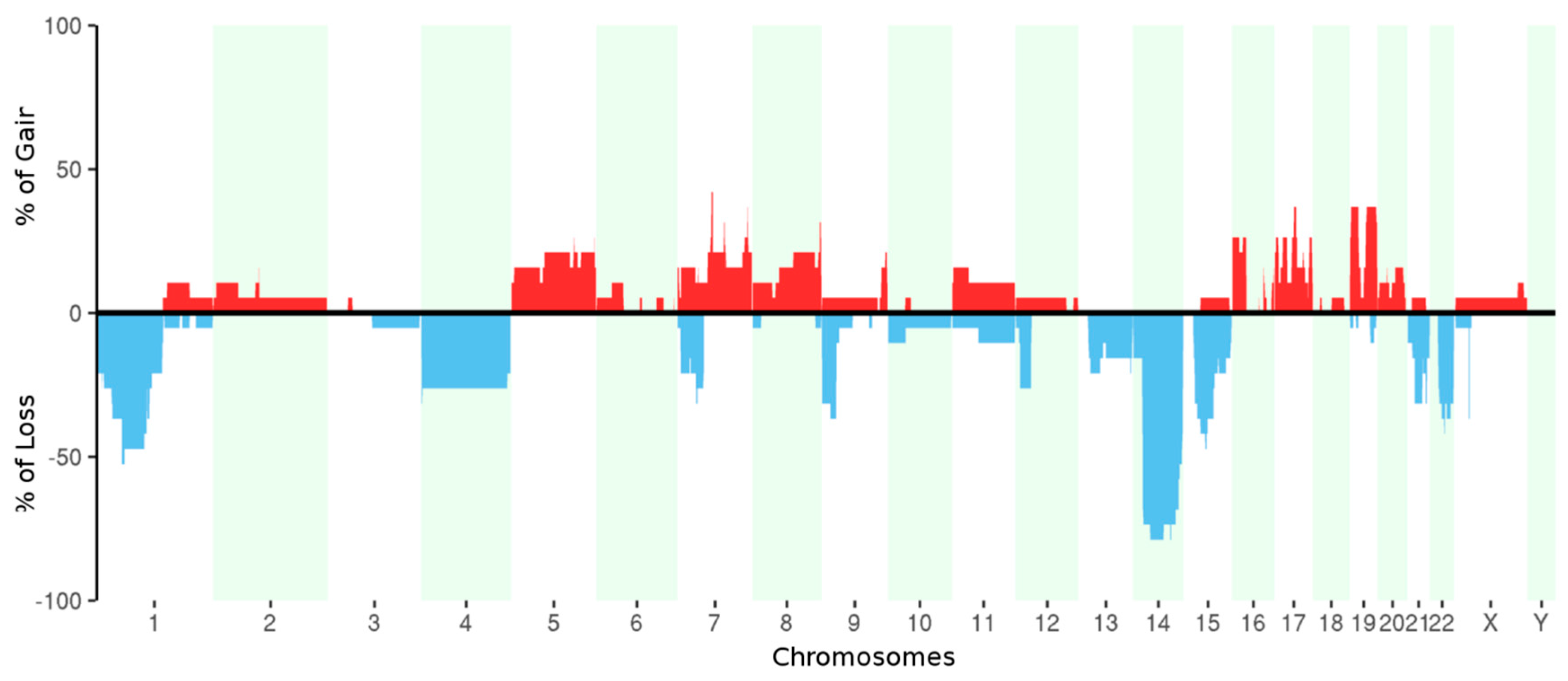
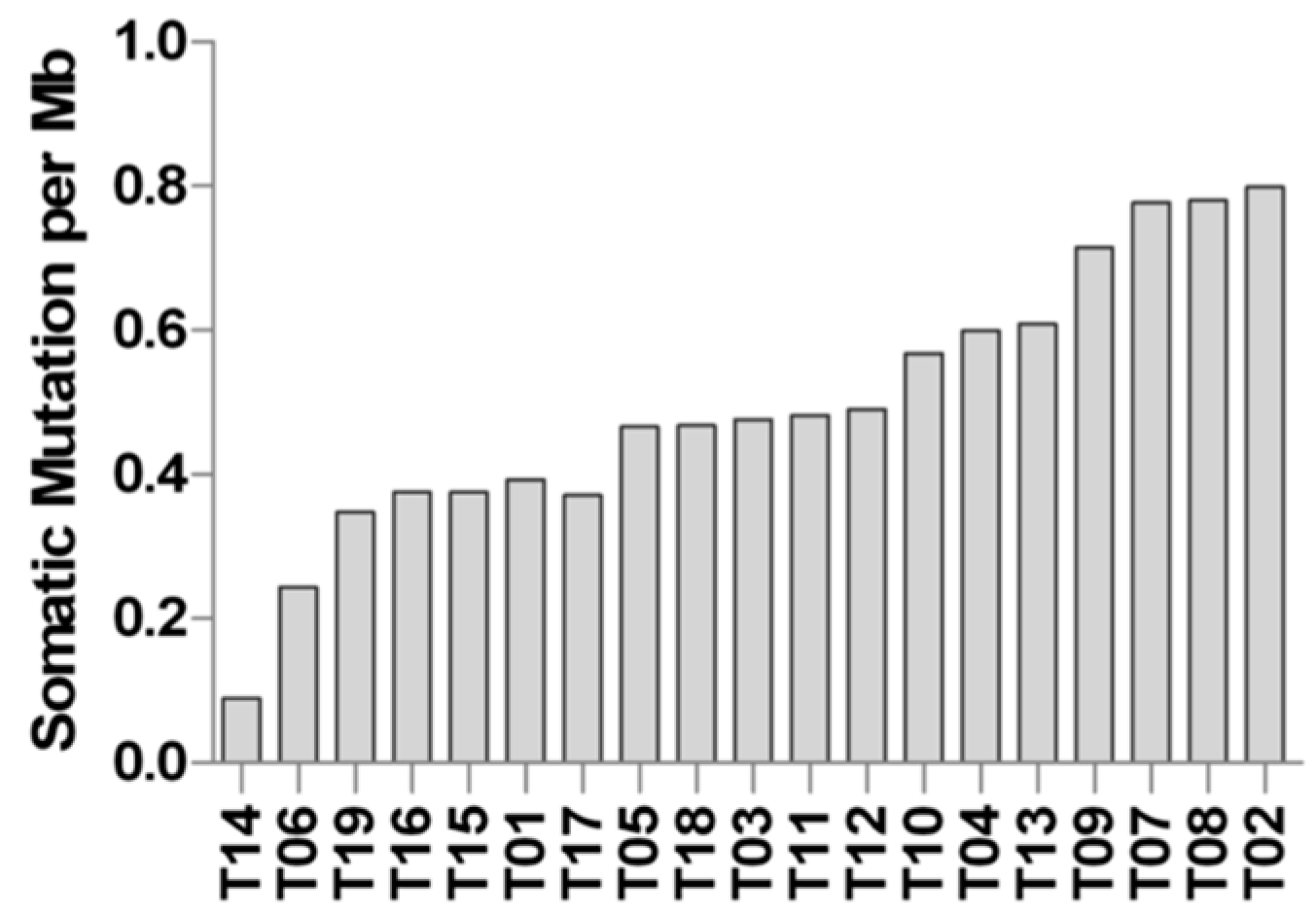
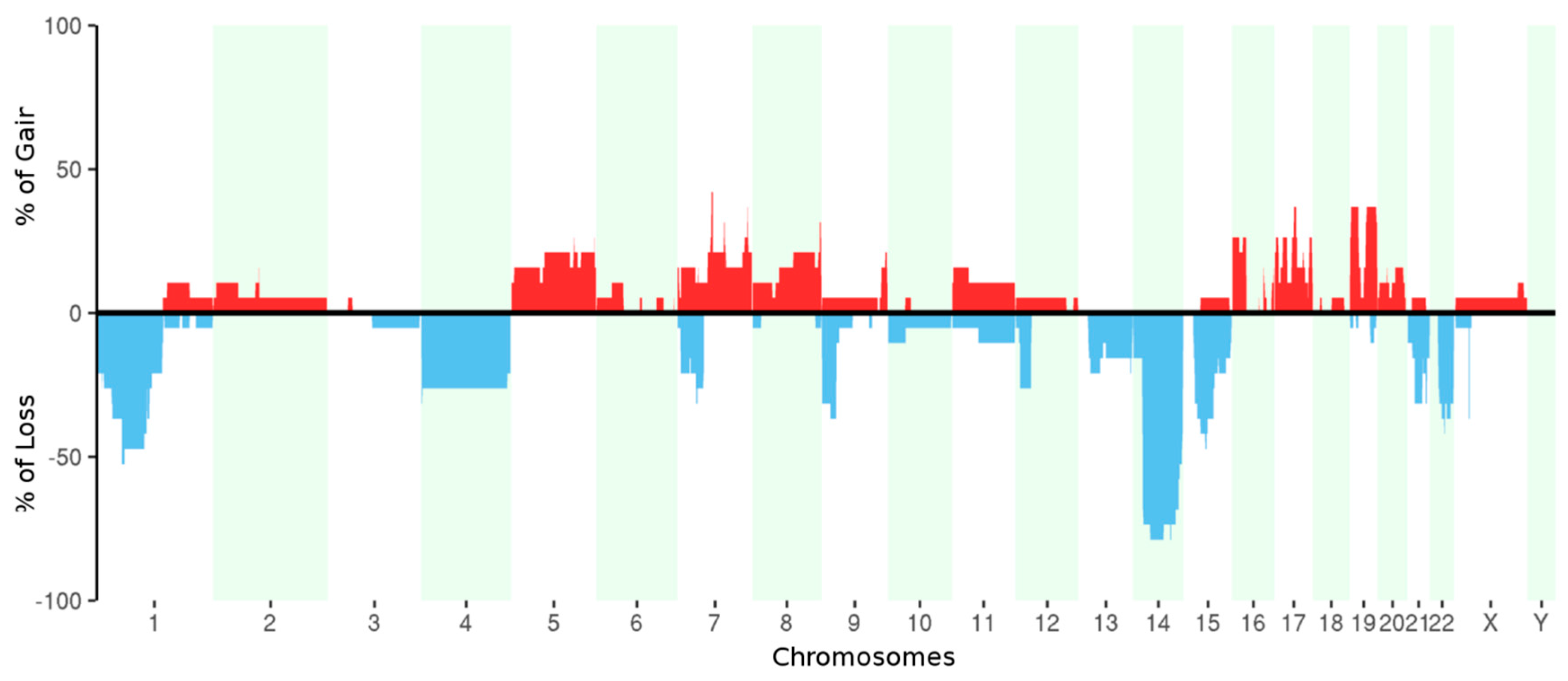
2.3. Whole Exome Sequencing
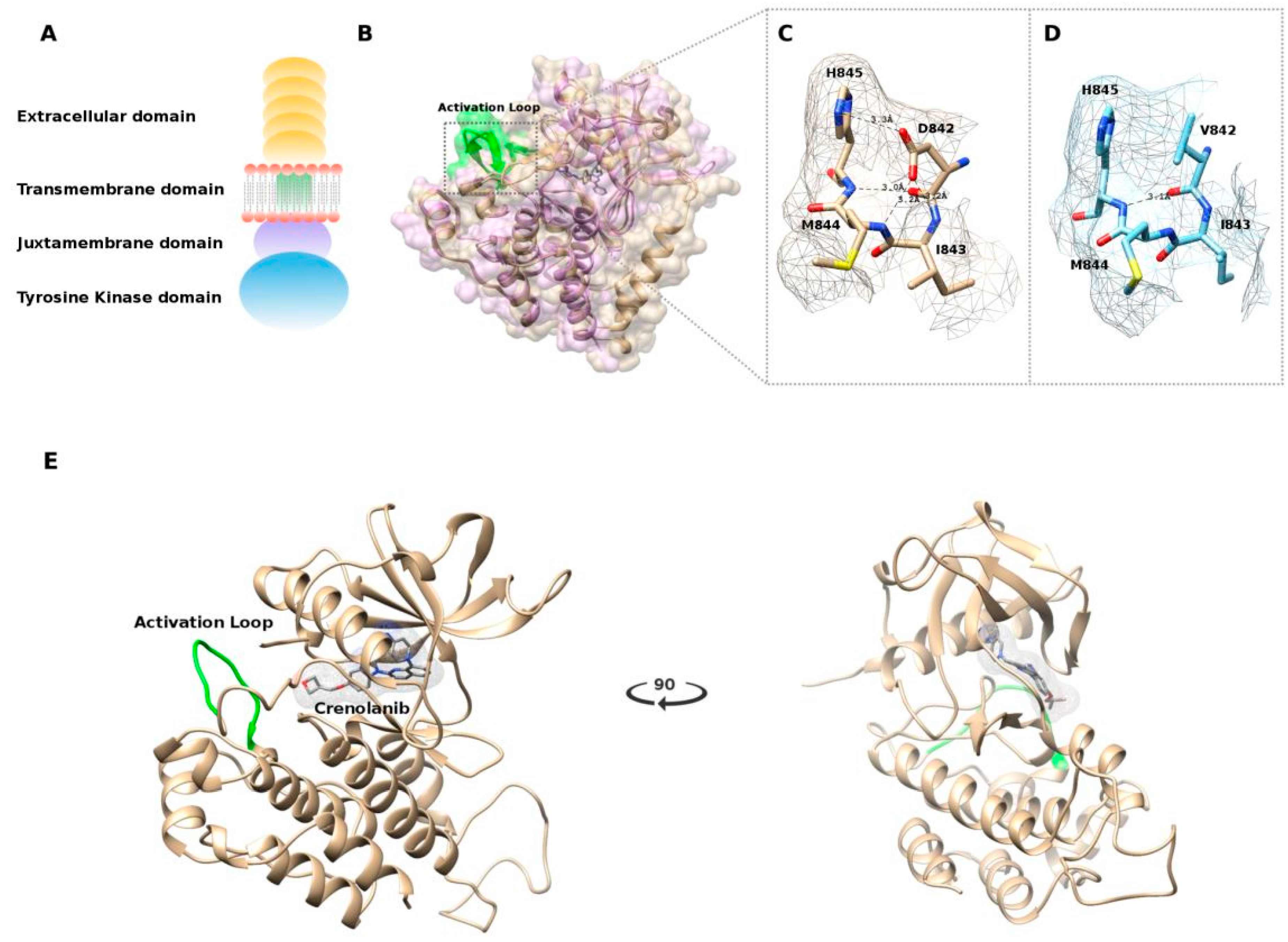
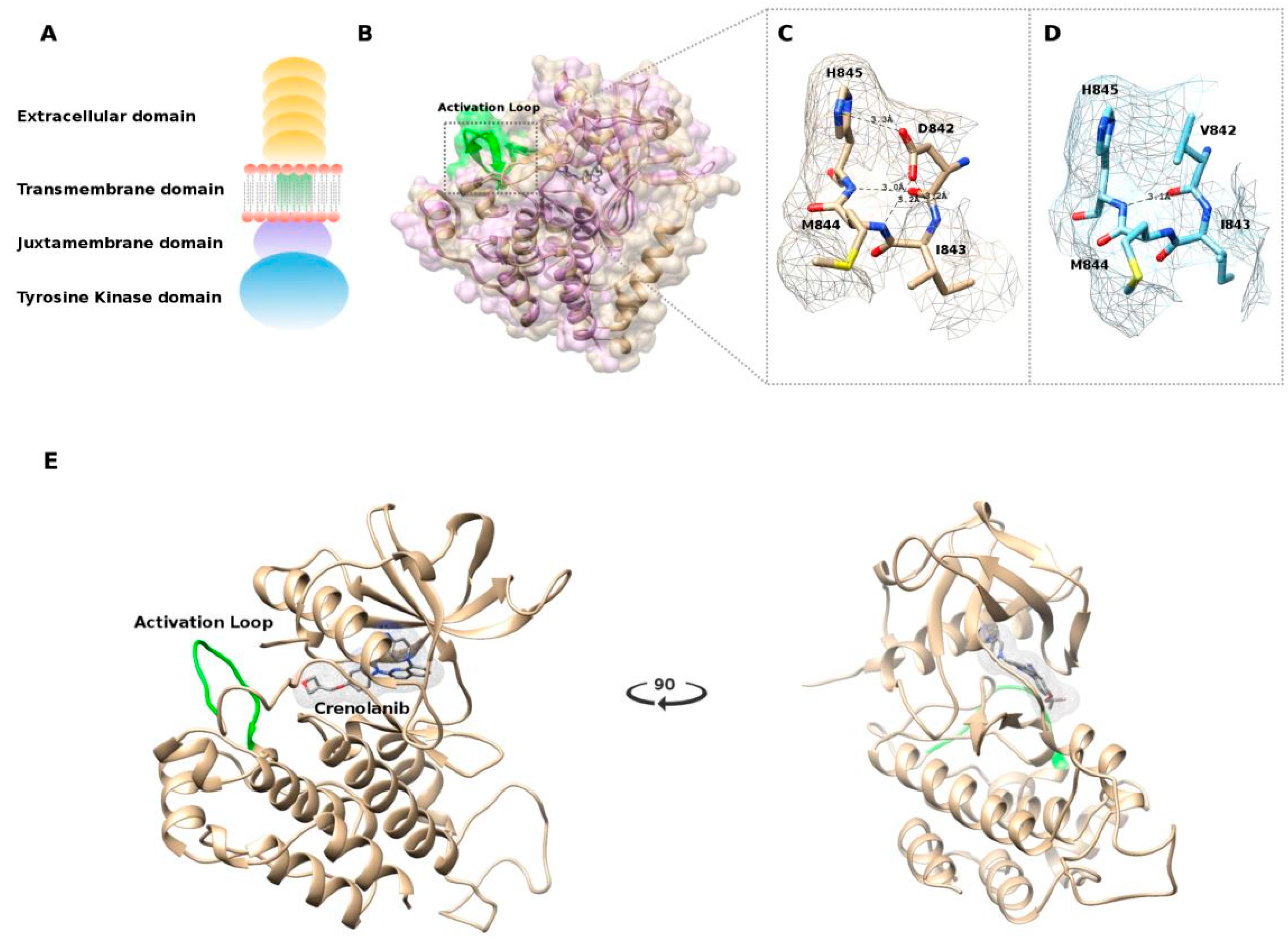
2.4. In Silico Modeling
3. Discussion
4. Materials and Methods
4.1. Whole-Transcriptome Paired-End RNA Sequencing and Whole Exome-Sequencing
4.2. Bioinformatic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Biasco, G.; Velo, D.; Angriman, I.; Astorino, M.; Baldan, A.; Baseggio, M.; Basso, U.; Battaglia, G.; Bertin, M.; Bertorelle, R.; et al. Gastrointestinal stromal tumors: Report of an audit and review of the literature. Eur. J. Cancer Prev. 2009, 18, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumors. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; Von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [PubMed]
- Debiec-Rychter, M.; Dumez, H.; Judson, I.; Wasag, B.; Verweij, J.; Brown, M.; Dimitrijevic, S.; Sciot, R.; Stul, M.; Vranck, H.; et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2004, 40, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1640 patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; Sciot, R.; Le, C.A.; Schlemmer, M.; Hohenberger, P.; Van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Cools, J.; Dumez, H.; Sciot, R.; Stul, M.; Mentens, N.; Vranckx, H.; Wasag, B.; Prenen, H.; Roesel, J.; et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005, 128, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Biron, P.; Cassier, P.A.; Fumagalli, E.; Blesius, M.; Debiec-Rychter, M.; Adenis, A.; Verweij, J.; Hohenberger, P.; Blay, J.; Casali, P.G.; et al. Outcome of patients (pts) with PDGFRA D842V mutant gastrointestinal stromal tumor (GIST) treated with imatinib (IM) for advanced disease. J. Clin. Oncol. 2010, 28, 10051. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib–resistant gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 4375–4384. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Bardsley, M.R.; Toyomasu, Y.; Milosavljevic, S.; Gajdos, G.B.; Choi, K.M.; Reid-Lombardo, K.; Kendrick, M.L.; Bingener-Casey, J.; Tang, C.M.; et al. Platelet-Derived Growth Factor Receptor-α Regulates Proliferation of Gastrointestinal Stromal Tumor Cells With Mutations in KIT by Stabilizing ETV1. Gastroenterology 2015, 149, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.L.; Lewis, L.D.; Eder, J.P.; Reddy, N.J.; Guo, F.; Pierce, K.J.; Olszanski, A.J.; Cohen, R.B. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J. Clin. Oncol. 2009, 27, 5262–5269. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Vlahovic, G.; Khamly, K.; Pierce, K.J.; Guo, F.; Olszanski, A.J. Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor. Br. J. Cancer 2010, 103, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.C.; Henze, J.; Fletcher, B.S.; Gu, Z.; et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Fuligni, F.; Saponara, M.; Nannini, M.; Lolli, C.; Indio, V.; Santini, D.; Ercolani, G.; et al. Dystrophin deregulation is associated with tumor progression in KIT/PDGFRA mutant gastrointestinal stromal tumors. Clin. Sarcoma Res. 2014, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Yan, X.E.; Yin, Y.; Yun, C.H. Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Biophys. Res. Commun. 2016, 477, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Lasater, E.A.; Lin, K.C.; Wang, Q.; McCreery, M.Q.; Stewart, W.K.; Damon, L.E.; Perl, A.E.; Jeschke, G.R.; Sugita, M.; et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 5319–5324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Zhuang, C.; Xu, J.; Wang, M.; Zhang, Z.Z.; Tu, L.; Wang, C.J.; Ling, T.L.; Cao, H.; Zhang, Z.G. Somatostatin receptors in gastrointestinal stromal tumors: New prognostic biomarker and potential therapeutic strategy. Am. J. Transl. Res. 2014, 6, 831–840. [Google Scholar] [PubMed]
- Kim, H.S.; Lee, H.S.; Kim, W.H. Clinical significance of protein expression of cyclooxygenase-2 and somatostatin receptors in gastroenteropancreatic neuroendocrine tumors. Cancer Res. Treat. 2011, 43, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, M.Z.; Wang, X.Q.; Tao, B.B.; Zhong, J.; Wang, X.H.; Zhang, W.C.; Li, S.T. MEM140 is associated with the prognosis of glioma by promoting cell viability and invasion. J. Hematol. Oncol. 2015, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Landin-Malt, A.; Benhaddou, A.; Zider, A.; Flagiello, D. An evolutionary, structural and functional overview of the mammalian TEAD1 and TEAD2 transcription factors. Gene 2016, 591, 292–303. [Google Scholar] [CrossRef] [PubMed]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, M.G.; Cai, K.Q.; Zhou, Y.; Luo, B.; Pei, J.; Rink, L.; Mehren, M. Succinate dehydrogenase deficiency in a PDGFRA mutated GIST. BMC Cancer 2017, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, D.; Rossi, S.; Campagna, D.; Scavina, P.; Tiziano, F.D.; Marzotto, A.; Toffolatti, L.; Vitelli, C.E.; Amini, M.; Dei Tos, A.P.; et al. Imatinib-Sensitizing KIT Mutation in a Carney-Stratakis-Associated GI Stromal Tumor. J. Clin. Oncol. 2016, 34, e99–e103. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Callender, G.G.; Kupferman, M.E.; Rich, T.A.; Chuang, H.H.; Trent, J.; Perrier, N.D.; Goodarzi, M.; Jimenez, C. Paraganglioma syndrome type 1 in a patient with Carney-Stratakis syndrome. Nat. Rev. Endocrinol. 2010, 6, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schoffski, P.; Bauer, S.; Mir, O.; Cassier, P.A.; Eskens, F.; Shi, H.; Alvarez-Diez, T.; et al. Clinical activity of BLU-285 in advanced gastrointestinal stromal tumor (GIST). J. Clin. Oncol. 2017, 35, 11011. [Google Scholar]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusiontranscripts. Genome Biol. 2011, 12, R72. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.; Griffith, M.; Moussavi, A.H.; Senz, J.; Melnyk, N.; et al. deFuse: An algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Chinnaiyan, A.M.; Maher, C.A. Chimera Scan: A tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011, 27, 2903–2904. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Liu, K.; Juan, T.; Fang, F.; Newman, M.; Hoeck, W. Fusion Map: Detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics 2011, 27, 1922–1928. [Google Scholar] [CrossRef] [PubMed]
- Nannini, M.; Astolfi, A.; Urbini, M.; Indio, V.; Santini, D.; Heinrich, M.C.; Corless, C.L.; Ceccarelli, C.; Saponara, M.; Mandrioli, A.; et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 2014, 14, 685. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, K.C.; Li, J.; Hunter, S.M.; Ryland, G.L.; Cowin, P.A.; Campbell, I.G.; Halgamuge, S.K. Inferring copy number and genotype in tumour exome data. BMC Genom. 2014, 15, 732. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protocol Bioinform. 2016. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDockTools4: Automated docking with selective receptorflexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughputcrystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 8, 1355–1363. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sample | Age at Surgery | Sex | Type of Sample | Primary Tumor Location | Tumor Size | Mitotic Index 50HPF | Source | Treatment Before Sampling | WES | RNA-Seq |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P01 | T01 | 62 | M | Frozen | stomach | 11 | 300 | metastasis | C, D | 101X1 | NA |
| P02 | T02 | 74 | F | Frozen | stomach | 16.2 | 5 | metastasis | C | 101X1 | NA |
| P03 | T03 | 51 | M | Frozen | stomach | 5 | 55 | metastasis | I, G/D, Su, IPI-504, So, STA-9090, D, GDC-0980, C | 101X1 | NA |
| P04 | T04 | 51 | M | FFPE | stomach | 6.2 | 7 | metastasis | none | 101X1 | NA |
| T05 | 51 | M | FFPE | stomach | 6.2 | 7 | metastasis | none | 101X1 | NA | |
| P05 | T06 | 53 | M | Frozen OCT | NA | NA | NA | primary | none | 101X1 | NA |
| P06 | T07 | 56 | M | FFPE | stomach | 30 | high | metastasis | I, Su, So, N | 101X1 | NA |
| T08 | 56 | M | FFPE | stomach | 30 | high | metastasis | I, Su, So, N | 101X1 | NA | |
| T09 | 56 | M | FFPE | stomach | 30 | high | metastasis | I, Su, So, N | 101X1 | NA | |
| T10 | 56 | M | FFPE | stomach | 30 | high | metastasis | I, Su, So, N | 101X1 | NA | |
| T11 | 56 | M | FFPE | stomach | 30 | high | metastasis | I, Su, So, N | 101X1 | NA | |
| P07 | T12 | 63 | F | Frozen | stomach | 10.5 | 19 | NA | C | 101X1 | NA |
| P08 | T13 | 76 | M | Frozen | stomach | NA | NA | primary | none | 101X1 | NA |
| P09 | T14 | 30 | M | Frozen | stomach | NA | NA | primary | none | 101X1 | NA |
| P10 | T15 | 50 | F | Frozen | stomach | 1.8 | 2 | primary | none | 100X2 | 75X2 |
| P11 | T16 | 75 | F | Frozen | stomach | 7 | 8 | primary | none | 100X2 | 75X2 |
| P12 | T17 | 68 | M | Frozen | stomach | 3 | 5 | primary | none | 100X2 | 75X2 |
| P13 | T18 | 76 | M | Frozen | stomach | 4.5 | 6 | primary | none | 100X2 | 75X2 |
| P14 | T19 | 51 | M | Frozen | stomach | 12 | 2 | primary | none | 100X2 | 75X2 |
| Pathway | Reactome Gene Set | Description | # Genes (Total) | # Leading-Edge Genes | NES | p Value | FDR |
|---|---|---|---|---|---|---|---|
| Positively related Pathways | R-HSA-373076 | Class A/1 (Rhodopsin-like receptors) | 30 | 24 | 2.03 | <0.001 | 0.0058 |
| R-HSA-375276 | Peptide ligand-binding receptors | 18 | 14 | 1.94 | 0.001 | 0.014 | |
| R-HSA-500792 | GPCR ligand binding | 38 | 27 | 1.90 | <0.001 | 0.01975 | |
| R-HSA-1630316 | Glycosaminoglycan metabolism | 19 | 13 | 1.77 | 0.001 | 0.09927 | |
| R-HSA-388396 | GPCR downstream signaling | 70 | 35 | 1.74 | 0.001 | 0.13397 | |
| R-HSA-3560782 | Diseases associated with glycosaminoglycan metabolism | 6 | 6 | 1.70 | 0.005 | 0.15244 | |
| R-HSA-1638091 | Heparan sulfate/heparin (HS-GAG) metabolism | 9 | 8 | 1.71 | 0.003 | 0.15294 | |
| R-HSA-372790 | Signaling by GPCR | 99 | 45 | 1.68 | <0.001 | 0.16662 | |
| R-HSA-1296072 | Voltage gated Potassium channels | 6 | 4 | 1.66 | 0.003 | 0.19534 | |
| R-HSA-112314 | Neurotransmitter Receptor Binding And Downstream Transmission In The Postsynaptic Cell | 17 | 10 | 1.65 | 0.014 | 0.19979 | |
| Negatively related Pathways | R-HSA-1640170 | Cell Cycle | 56 | 49 | −2.83 | <0.001 | <0.001 |
| R-HSA-69242 | S Phase | 15 | 15 | −2.53 | <0.001 | 0.00088 | |
| R-HSA-69278 | Cell Cycle, Mitotic | 41 | 37 | −2.57 | <0.001 | 0.00131 | |
| R-HSA-2559583 | Cellular Senescence | 13 | 13 | −2.46 | <0.001 | 0.00162 | |
| R-HSA-69239 | Synthesis of DNA | 14 | 14 | −2.47 | <0.001 | 0.00202 | |
| R-HSA-69306 | DNA Replication | 14 | 14 | −2.42 | <0.001 | 0.00266 | |
| R-HSA-69481 | G2/M Checkpoints | 18 | 17 | −2.29 | <0.001 | 0.00723 | |
| R-HSA-73894 | DNA Repair | 29 | 28 | −2.22 | <0.001 | 0.00920 | |
| R-HSA-5693538 | Homology Directed Repair | 16 | 15 | −2.21 | <0.001 | 0.01075 | |
| R-HSA-5693532 | DNA Double-Strand Break Repair | 16 | 15 | −2.18 | <0.001 | 0.01076 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Indio, V.; Astolfi, A.; Tarantino, G.; Urbini, M.; Patterson, J.; Nannini, M.; Saponara, M.; Gatto, L.; Santini, D.; Do Valle, I.F.; et al. Integrated Molecular Characterization of Gastrointestinal Stromal Tumors (GIST) Harboring the Rare D842V Mutation in PDGFRA Gene. Int. J. Mol. Sci. 2018, 19, 732. https://doi.org/10.3390/ijms19030732
Indio V, Astolfi A, Tarantino G, Urbini M, Patterson J, Nannini M, Saponara M, Gatto L, Santini D, Do Valle IF, et al. Integrated Molecular Characterization of Gastrointestinal Stromal Tumors (GIST) Harboring the Rare D842V Mutation in PDGFRA Gene. International Journal of Molecular Sciences. 2018; 19(3):732. https://doi.org/10.3390/ijms19030732
Chicago/Turabian StyleIndio, Valentina, Annalisa Astolfi, Giuseppe Tarantino, Milena Urbini, Janice Patterson, Margherita Nannini, Maristella Saponara, Lidia Gatto, Donatella Santini, Italo F. Do Valle, and et al. 2018. "Integrated Molecular Characterization of Gastrointestinal Stromal Tumors (GIST) Harboring the Rare D842V Mutation in PDGFRA Gene" International Journal of Molecular Sciences 19, no. 3: 732. https://doi.org/10.3390/ijms19030732