The Target of Rapamycin and Mechanisms of Cell Growth


Division of Cancer and Genetics, Cardiff University, Heath Park, Cardiff CF14 4XN, UK
Int. J. Mol. Sci. 2018, 19(3), 880; https://doi.org/10.3390/ijms19030880
Submission received: 19 February 2018
/
Revised: 12 March 2018
/
Accepted: 13 March 2018
/
Published: 16 March 2018
(This article belongs to the Special Issue Cell Growth Regulation)
{kind=link}
Abstract
:Mammalian target of rapamycin (mTOR, now referred to as mechanistic target of rapamycin) is considered as the master regulator of cell growth. A definition of cell growth is a build-up of cellular mass through the biosynthesis of macromolecules. mTOR regulation of cell growth and cell size is complex, involving tight regulation of both anabolic and catabolic processes. Upon a growth signal input, mTOR enhances a range of anabolic processes that coordinate the biosynthesis of macromolecules to build cellular biomass, while restricting catabolic processes such as autophagy. mTOR is highly dependent on the supply of nutrients and energy to promote cell growth, where the network of signalling pathways that influence mTOR activity ensures that energy and nutrient homeostasis are retained within the cell as they grow. As well as maintaining cell size, mTOR is fundamental in the regulation of organismal growth. This review examines the complexities of how mTOR complex 1 (mTORC1) enhances the cell’s capacity to synthesis de novo proteins required for cell growth. It also describes the discovery of mTORC1, the complexities of cell growth signalling involving nutrients and energy supply, as well as the multifaceted regulation of mTORC1 to orchestrate ribosomal biogenesis and protein translation.
1. Introduction
1.1. History of Rapamycin and Drug Targets
The background history of the discovery of mechanistic target of rapamycin (mTOR) is atypical. More commonly, small molecule kinase inhibitors are developed after discovery of the protein kinase. However, in the case of mTOR, this protein kinase was discovered through the drug activity of a naturally occurring inhibitor called rapamycin. Rapamycin (also known as sirolimus and later marketed under the trade name Rapamune by Pfizer) is a macrocyclic lactone isolated from Streptomyces hygroscopicus, a bacterium extracted from a soil sample on Easter Island (known as ‘Rapa-Nui’) [1]. The biosynthesis of rapamycin is an energy intensive multi-step process involving many multifunctional enzymes (reviewed in [2]). The bacteria acquire a selective advantage by synthesising and secreting rapamycin into the soil. Rapamycin has antifungal properties, enabling the bacteria to more easily colonise the soil by repressing the growth of competing fungi [3]. Originally defined as an antifungal compound in the mid-70s, rapamycin was later found to be effective as an immunosuppressant with anti-proliferative properties in humans [4,5].
While the drug property of rapamycin to impair growth was beginning to be realised, the drug target was unknown until the early 1990s. The first breakthrough into the mechanism of drug activity was the finding that rapamycin interacted with FKBP12 (FK506-binding protein 12), an immunophilin in mammalian cells (reviewed in [6]). Following on from this, research in yeast led to a series of pivotal discoveries. By using FK506 columns, the yeast FKBP (FK506-binding protein) was purified and was identified as FK506-binding protein 1 (FRR1) [7]. Surprisingly, yeast genetics revealed that FRR1 was not involved in controlling cell growth. This was because genetic loss of FRR1 did not arrest growth like rapamycin, implying that there had to be another drug target of rapamycin [4,7,8]. To identify this elusive drug target, genetic screens in yeast were carried out for rapamycin-resistant mutants [7,9]. These genetic screens did not only reveal FRR1, but also discovered two genes that were accordingly named as target of rapamycin 1 (TOR1) and TOR2. Disruption of both TOR1 and TOR2 phenocopied rapamycin to cause cell growth arrest in yeast [10]. From these early studies, it was ascertained that rapamycin acted on TOR1 and TOR2 to repress cell growth. However, the other piece of the puzzle involving rapamycin-FRR1 (as a drug-immunophilin complex) was not yet recognised. For some drug-immunophilin complexes, drug association with an immunophilin results in adding an additional drug activity, which is observed when cyclosporine A interacts with FK506 [6]. Therefore, it was speculated that rapamycin might gain a new drug activity to arrest cell growth when associated with an FKBP. Substantiating this idea, the rapamycin-FRR1 complex in yeast was later found to bind directly to TOR1 and TOR2 to arrest cell growth [11,12,13]. In mammalian systems, it was also observed that a homologous protein to yeast TOR1 and TOR2 directly interacted with FKBP12-rapamycin [14,15], which was later called mTOR. By the mid-1990’s, the drug activity of rapamycin was better understood; rapamycin could bind to and inhibit TOR as a rapamycin-FKBP drug-immunophilin complex. In yeast, loss of TOR1 and TOR2 phenocopies nutrient starvation to cause inhibition of protein synthesis, glycogen accumulation and autophagy induction [16,17,18,19]). Since these early yeast studies, much has been discovered, including delineation of the mTOR signalling network and conservation of this pathway in higher eukaryotic cells.
1.2. mTOR Structure and Protein Complexes
mTOR is a member of the phosphatidylinositol 3-kinase-related kinases (PIKK) family, which includes DNA-dependent protein kinase (DNA-PK, also known as protein kinase, DNA-activated, catalytic polypeptide), ATM serine/threonine kinase (ATM) and ATR serine/threonine kinase (ATR). Instead of having lipid kinase activity, mTOR’s kinase catalytic domain functions to phosphorylate proteins on Ser/Thr residues [15,20]. Distinguishing features of mTOR include multiple HEAT (Huntington, EF3, A subunit of PP2A, TOR1) repeats within the N-terminus that permits multiple protein-protein interactions [21]. mTOR also possesses a central FAT (FRAP, ATM, TRAP) domain and a C-terminal FAT (FATC) domain that are also conserved in other PIKK family members [22]. Allosteric interaction of FKBP12-rapamycin with the FKBP-rapamycin binding (FRB) domain is inhibitory to the phosphotransferase activity of mTOR. The FRB domain is situated between the central FAT and the kinase domain [11,14].
While yeast possesses two TOR genes, there is only one mTOR gene in higher eukaryotes. Rather than gene duplication as an evolutionary route to generate two distinctive TOR protein kinases as observed in yeast, higher eukaryotes instead possess a single mTOR gene that becomes integral to two protein kinase complexes, called mTOR complex 1 (mTORC1) and mTORC2. Both mTORC1 and mTORC2 are high molecular weight protein complexes that contain an array of core binding and regulatory proteins that are associated with the mTOR catalytic subunit. mTORC1 was first identified by the association of mTOR with rapamycin-associated protein of TOR (Raptor) [23,24] and MTOR associated protein, LST8 homolog (mLST8) [25]. Raptor functions as a scaffold protein and it’s binding to mTOR is necessary for mTORC1 substrate specificity (reviewed in [26]). mTORC1 also interacts with regulatory proteins that inhibit mTORC1, proline-rich Akt substrate of 40 kDa (PRAS40) [27,28,29] and DEP domain containing mTOR-interacting protein (DEPTOR) [30]. Other regulatory mTORC1 interacting proteins include the telomere maintenance 2 (TELO2) and TELO2 interacting protein 1 (TTI1), which associates with mTORC1 as a TELO2/TTI1 complex and is necessary for mTORC1 assembly [31]. For mTORC2, mLST8 and DEPTOR also interact. Core binding subunits that are exclusive to mTORC2 include rapamycin-insensitive companion of mammalian target of rapamycin (Rictor), mSin1 (mammalian stress-activated protein kinase interacting protein 1) and Protor (protein observed with Rictor-1) [32,33]. Rictor is essential for substrate specificity of mTORC2 as well as its assembly and stability, while mSin1 functions as a negative regulator (reviewed in [34]). mTORC2 is a regulator of the cytoskeleton through its stimulation of F-actin stress fibers. mTORC2 directly phosphorylates and activates AKT to enhance cell growth via mTORC1. As mTORC1 is more centrally involved in the control of cell growth when compared to mTORC2, this review focuses mainly on mTORC1-regulated processes.
mTORC1 and mTORC2 can be distinguished by their differences in rapamycin sensitivity. Prior to the discovery of the two mTOR complexes, it was commonly presumed that rapamycin was effective at blocking the kinase activity of mTOR. However, the kinase activity of mTORC2 is insensitive to rapamycin, which was the principle reason why mTORC2-driven processes remained hidden in earlier studies that employed rapamycin. Furthermore, it is now appreciated that some mTORC1-mediated phosphorylation events are partially resistant to rapamycin [35,36]. As an example, mTORC1-mediated phosphorylation of a well-characterised mTORC1 substrate, eukaryotic initiation factor-4E-binding protein 1 (4E-BP1) at Thr37/Thr46 is heavily resistant to rapamycin treatment [37]. Another partially rapamycin resistant process that is regulated by mTORC1 is autophagy, where the level of resistance varies depending on cell-type [36]. It was hypothesised that cells with a more open (or relaxed) mTORC1 conformation would likely have higher sensitivity to rapamycin [36]. For mTORC2, short-term rapamycin treatment is not inhibitory. However, longer durations of treatment can indirectly inhibit mTORC2. This is because rapamycin-FKBP12 associates with newly synthesised mTOR prior to complex assembly, sequestering this “free pool” of mTOR from Rictor to prevent mTORC2 formation. Consequently, prolonged treatments of rapamycin (over days of treatment) can indirectly lead to reduced mTORC2 activity [38].
Rapamycin has been instrumental in the discovery of mTOR and has helped elucidate rapamycin sensitive cell processes that were later found to be driven by mTORC1. However, with the introduction of gene silencing and targeted genomic editing technologies, a much greater appreciation of mTORC1 and mTORC2 is beginning to emerge, where knockdown or knockout of either Raptor or Rictor is now more frequently used to ablate mTORC1 or mTORC2 signal transduction, respectively. Furthermore, the introduction of ATP-competitive inhibitors of mTOR adds an additional research tool, allowing researchers to more fully repress the kinase activity of both mTOR complexes.
2. mTORC1 and Cell Growth Control
2.1. Upstream Signalling Pathway of mTORC1
Model systems such as Drosophila have been instrumental in understanding the role that mTOR has in cell growth control and organismal size. Genetic analysis of the Drosophila TOR homolog, dTOR (also now being referred to as mTOR), clearly revealed that inactivating mTOR mutations cause a delay in cell proliferation and reduce cell size [39]. While conversely, mTOR activation promoted cell and organ size [39]. Another key discovery made was the identification of the Drosophila genes dTSC1 (Tuberous sclerosis complex 1) and dTSC2 as regulators of cell size [40,41,42]. Genetic epistasis experiments uncovered that dTSC1 and dTSC2 functioned as negative regulators of cell size, positioning them downstream of the insulin receptor and AKT (AKT serine/threonine kinase) [40,41]. These genetic studies positioned Drosophila ribosomal protein S6 kinase (dS6K) downstream of dTSC1 and dTSC2. In fruit flies, dS6K controls cell size, where dS6K inactivation mutations cause a small fly phenotype [43]. dS6K, as well the mammalian homologue, ribosomal protein S6 kinase 1 (S6K1) are well-characterised rapamycin-sensitive substrates of mTORC1 [44]. These fly studies inferred that dTSC1 and dTSC2 functioned upstream of mTORC1. Sequential studies confirmed that dTSC1/dTSC2 were genetically positioned upstream from mTOR. Inactivation mutations of mTOR blocked the large cell phenotype in dTsc2 mutant flies [45]. Furthermore, mutations reducing the activity of mTOR and dS6K rescued the lethality caused by dTSC1 mutations [42]. Therefore, it was discovered that dTSC1 and dTSC2 functioned to repress growth signals upstream of mTORC1.
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic condition caused by loss of function mutations in either TSC1 or TSC2. TSC1 and TSC2 are tumour suppressors that function together, where loss of function mutations of either gene predisposes the growth of tumours in multiple organs in TSC patients, including the kidney, brain and skin, as well as neurocognitive problems and epilepsy [46]. Tumour growth in TSC patients is now known to be dependent on mTORC1. Consequently, primary care to treat TSC patients include the use of mTORC1 inhibitors, such as everolimus [46]. Basic research showing the involvement of TSC1/TSC2 in mTORC1 signal transduction followed by rapamycin pre-clinical studies in mouse models of TSC [47] were instrumental in the repositioning of mTORC1 inhibitors to treat TSC. Basic research that followed on from the genetic analysis in Drosophila described above revealed a conserved pathway in mammals. Within the phosphatidylinositide 3-kinase (PI3K)/AKT signalling pathway, AKT directly phosphorylates TSC2 on four or five residues, revealing that TSC1/TSC2 is directly regulated by AKT [48,49,50]. Over-expression of TSC1 and TSC2 also markedly inhibited S6K1, positioning TSC1/TSC2 upstream of S6K1. TSC1/TSC2 was then later shown to repress signal transduction through mTORC1 [51]. It was assumed that a small-G protein was likely regulated by TSC1/TSC2, as TSC2 (also known as tuberin) possessed a conserved C-terminal GTPase activating protein (GAP) domain that was commonly lost in TSC patients (via C-terminal truncating point-nonsense mutations) [52,53]. Furthermore, a cluster of pathogenic single point mutations within the conserved GAP domain of TSC2 was also reported [53]. In yeast, a likely small G-protein candidate was Ras homologue enriched in brain (Rheb), where genetic loss of Rheb in yeast phenocopied nutrient starvation [54]. Confirming the involvement of this small G protein, Rheb was found to be regulated by TSC1/TSC2 in mammalian cells [55,56,57]. When complexed with TSC1, TSC2 functions as a Rheb GAP, switching Rheb to an inactive GDP-bound form to turn mTORC1 off [57]. Rheb becomes activated, i.e., GTP-loaded, when the TSC1/TSC2 complex is repressed through an array of upstream kinases within mitogenic and hormone stimulated pathways (depicted in Figure 1). Paralleling the PI3K/AKT pathway, mitogen-activated protein kinases (MAPK) also converges on TSC1/TSC2 to positively regulate mTORC1 [58]. It was later found that activation of MAPK also inhibits TSC1/TSC2 through TSC2 phosphorylation by RSK [59] and ERK [60].
2.2. Regulation of Protein Translation and Cell Growth by mTORC1
mTORC1 relays nutrient, energy and growth signals to drive cell growth through promotion of anabolic processes such as protein synthesis. As well as enhancing the efficiency of protein translation, mTORC1 promotes protein translation through increasing the production of ribosomes. Furthermore, mTORC1 boosts the generation of nucleotide precursors that are essential for a growing cell. These nucleotide precursors are required for the generation of rRNA to build new ribosomes, mRNA to be transcribed into proteins necessary for growth and dNTP nucleotides for DNA replication during cell division.
mTORC1 regulates protein translation through an array of translation factors that include 4E-BP1 and S6K1. Both 4E-BP1 and S6K1 possess an mTORC1 signalling (TOS) motif, which associates with Raptor [61]. Consequently, the TOS motif is necessary for the recruitment of both 4E-BP1 and S6K1 to mTORC1 and the phosphorylation of these substrates [61]. 4E-BP1 acts as a negative regulator of cap-dependant protein translation. eukaryotic initiation factor (eIF) 4E associates with the m7GpppN cap moiety on the 5′-end of mRNAs and is inactivated through association with 4E-BP1 when in an unphosphorylated state, thus preventing the translation of mRNAs involved in cell growth [62]. mTORC1 phosphorylates 4E-BP1 on four Ser/Thr residues, causing dissociation of 4E-BP1 from eIF4E. As association of either 4E-BP1 or eIF4G to eIF4E are mutually exclusive, eIF4G then associates with eIF4E to promote translation initiation (reviewed in [21]). eIF4G acts as a scaffold to recruit other translation initiation factors, such as eIF4A, to form the eIF4F complex. Assembly of eIF4F is considered a rate-limiting step of translation initiation. As part of the eIF4F complex, eIF4A has RNA helicase activity that unwinds the secondary structure within the 5′-untranslated region (5′-UTR) of the mRNA to help the efficacy of ribosome scanning to the start codon. Via mTORC1, S6K1 further enhances the RNA helicase activity of eIF4A by phosphorylating eIF4B on Ser422 [63]. mRNAs involved in cell growth are heavily dependent on mTORC1. The length and degree of secondary structure within the 5’-UTR contributes to the dependency of these mRNAs to mTORC1 and the availability of eIF4F (reviewed in [64]). Examples of mRNAs involved in cell growth that are highly dependent on eIF4F include the MYC proto-oncogene, bHLH transcription factor (MYC) and cyclin D1 (CCND1) [65].
2.3. Ribosomal Biogenesis and mTORC1
The cells’ capacity to generate protein is limited by the number of ribosomes they have. Consequently, mTORC1 promotes ribosomal biogenesis to ensure rapid growth. Typically, 5′-terminal oligopyrimidine (5′-TOP) tracts are found in mRNA that encode for factors involved in ribosomal biogenesis and ribosomal proteins [66,67]. These 5′-TOP tracts function as translational cis-regulatory elements. 5′-TOP mRNAs are sensitive to rapamycin and are found to be dependent on S6K1 [68]. Over 75% of the proteins involved in ribosomal biogenesis are estimated to be controlled by S6K1 [68]. In a recent study, high resolution ribosomal profiling identified 144 mRNAs that were sensitive to mTORC1 inhibitors [69]. Only 68% of these mRNAs possessed putative 5′-TOP tracts, while 63% contained a newly discovered cis-regulatory pyrimidine-rich translational element (PRTE). Rather than being dependent on S6K1, PRTE mRNAs were found to be highly sensitive to 4E-BP1. Many of the mTORC1-sensitive target genes uncovered by Hsieh et al., were ribosomal proteins [69], which highlights the critical involvement of mTORC1 in the generation of new ribosomes. The regulatory mechanism of these translational cis-regulatory elements is currently unknown. Presumably, trans-acting factors bind to these elements and regulate the translation of the mRNA. Several potential trans-acting factors that bind to the 5′-TOP have been studied to date that include the La protein [70] and the La-related proteins (LARP) [71,72,73].
mTORC1 also promotes the generation of rRNA. rRNAs are the major component of the ribosome that comprise of 60% of the ribosomal mass. Eukaryotic ribosomes consist of four rRNAs called the 5S, 5.8S, 18S and 28S. RNA polymerase I (Pol I) transcribes a precursor 47S pre-rRNA that is sequentially processed in the nucleoli to form the mature 5.8S, 18S and 28S rRNA species. The 5S rRNA is generated by Pol III, which is also involved in the formation of transfer RNA (tRNA). To further enhance the production of rRNA, mTORC1 activates both Pol I and Pol III via several mechanisms. mTORC1 directly phosphorylates Transcription Initiation Factor I (TIF1A) to cause its translocation to the nucleolus and activation of Pol I [74]. mTORC1 further promotes Pol I via S6K1. S6K1 phosphorylates Upstream Binding Factor (UBF), causing UBF-TIF1B interaction and Pol I activation [75]. Through Pol I, mTORC1 and S6K1 enhance the production of 5.8S, 18S and 28S rRNA. mTORC1 also enhances the production of the 5S rRNA species by phosphorylating and inhibiting a negative regulator of Pol III, called MAF1 (MAF1 homolog, negative regulator of RNA polymerase III) [76]. In another study, mTORC1 was shown to directly bind to the promoter region of Pol I and Pol III, leading to their enhanced gene-expression [77]. Therefore, mTORC1-dependent regulation of ribosomal proteins and rRNA during ribosomal biogenesis is multifaceted.
To ensure that there is a sufficient supply of nucleotides for the generation of rRNA, mTORC1 promotes the production of nucleotide precursors. mTORC1 does this by redirecting glucose metabolites into the pentose phosphate pathway. The pentose phosphate pathway generates nicotinamide adenine dinucleotide phosphate, ribose-5-phosphate and erythrose-4-phosphate, precursors for fatty acids, nucleotides and aromatic amino acids, respectively. Generation of fatty acids, nucleotides and aromatic amino acids are essential for a growing cell, i.e., for the expansion of membranes and the de novo synthesis of mRNA, rRNA, DNA, and proteins. Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) within the pentose phosphate pathway was found to be phosphorylated and activated by S6K1 [78]. CAD catalyses the first three enzymatic steps of a six-step pyrimidine biosynthesis pathway to enrich the pyrimidine nucleotide pool [78]. More recently, mTORC1 was found to enhance purine biosynthesis through ATF4-mediated gene-expression of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), a metabolic enzyme involved in the synthesis of purine nucleotides [79]. In this study, it was found that mTORC1 increased the protein translation of ATF4, the transcription factor that drives the gene-expression of MTHFD2.
3. Nutrient and Energy Homeostasis during Cell Growth
Cell growth is tightly controlled by both nutrient and energy supply. If the supply of either nutrients or energy is insufficient, feedback mechanisms ensure that mTORC1 is switched off. Such signalling mechanisms maintain energy and nutrient homeostasis during cell growth.
3.1. Nutrient Signalling and mTORC1
Early studies implicated that mTORC1 required the presence of branch-chained amino acids for its full activation (reviewed in [80]). However, it was unclear how nutrients were “sensed” by mTORC1. The first indication that mTORC1 sensed an intracellular amino acid pool were in experiments that utilised cycloheximide, a drug that blocks translation elongation [81]. The amino acid reserves of a cell can become quickly exhausted via protein translation. Therefore, inhibition of protein translation with cycloheximide can be used to help indirectly replenish the intracellular amino acid stores. It was found that increasing the pool of intracellular amino acids after cyclohexmide treatment was sufficient to activate mTORC1 in the absence of external amino acid supply or growth factor stimuli [81]. It is now appreciated that intracellular nutrient sensing occurs at the level of lysosomes. In summary, Rag GTPase heterodimers regulate the localisation of mTORC1 to lysosomes [82,83]. During amino acid sufficiency, active Rag heterodimers (consisting of either RagA-GTP or RagB-GTP bound to either RagC-GDP or RagD-GDP) will associate with Raptor. These active Rag heterodimers bound to Raptor then translocate mTORC1 to the “Ragulator complex” found on the membrane surface of lysosomes, which is necessary for mTORC1 activation [84]. Under conditions of nutrient withdrawal, mTORC1 is cytoplasmically localised and is inactive. The Rag proteins are regulated by the Ragulator complex that is comprised of five proteins referred to as the late endosomal/lysosomal adaptor, MAPK and mTOR activator 1-5 (LAMTOR1-5) [84]. The Ragulator complex acts as a guanine nucleotide exchange factor (GEF) towards RagA and RagB, switching them to an active GTP-bound state [85]. RagA and RagB are negatively regulated by RagGTPases and GTRs-1 (GATOR) and GATOR2, that are also lysosomally localised [86]. GATOR1 acts as a RagGAP, switching both RagA and RagB to an inactive GDP-bound state to prevent lysosomal localisation of mTORC1 (to turn off mTORC1 signalling). GATOR2 lies directly upstream of GATOR1 and functions as a negative regulator of GATOR1. GATOR2 is positively regulated by nutrients [86].
Leucine was proposed to be sensed by three sestrins (SESN1-3) that activate GATOR2, which then in turn inactivates GATOR1 to switch the Rag heterodimers to an active state and promote mTORC1 signalling [87,88]. However, there is some controversy regarding SESN1-3 as a direct leucine sensor (reviewed in [89]). This is due to the known function of SESN1-3 to activate AMPK during cell stress [90,91,92]. Activation of AMPK can indirectly turn off mTORC1 (via a signalling feedback mechanism involving TSC1/2 that is summarised in Section 3.2 below). Indeed, genetic evidence in SESN-deficient fly and mouse models infer that the overriding function of SESN is to activate AMPK and to inhibit mTORC1, which importantly can occur in the presence of physiological levels of leucine [90,91,92]. It is possible that leucine is sensed by Leucyl-tRNA synthetase at the lysosome instead, which has been shown to function as a GAP towards RagD [93].
Arginine is sensed by mTORC1 via several mechanisms. It was recently found that TSC1/2 associates with Rheb when arginine levels are low, which causes TSC1/2 to switch Rheb to an inactive GDP-bound state to inhibit mTORC1 [94]. As another mechanism of arginine sensing, cytosolic arginine sensor for mTORC1 (CASTOR) subunit 1 interacts with GATOR2 in the absence of arginine to prevent mTORC1 activation [95]. When arginine levels are in sufficient supply, arginine binds to CASTOR1 to displace GATOR2 from the inhibited CASTOR1-CATOR2 complex. Consequently, GATOR2 is then able to turn off GATOR1, leading to mTORC1 activation [95]. Further studies in animal models will be required to validate these nutrient sensing mechanisms.
Evidently, nutrient sensing by mTORC1 is complex and involves multiple inputs. In the context of growth control, leucine is well known to play a significant role in the determination of muscle mass involving mTORC1. Leucine accounts for about 20% of our dietary protein intake and branch-chained amino acids account for about a third of muscle protein [96]. Muscle is the primary reservoir of protein within the human body. Therefore, it is not surprising that muscle cells are subjected to particularly high levels of protein turnover rates that help maintain amino acid homeostasis within the whole body. Leucine is critical for driving muscle growth and does this, in part, through the activation of mTORC1. Resistance exercise with protein supplements (including leucine) can promote the build-up of muscle mass [97]. Through activation of mTORC1, leucine is a potent activator of protein synthesis that drives anabolic muscle growth.
Nutrient withdrawal is known to restrict organ size and implicates the mTORC1 pathway and nutrient sensing in organismal growth. Growth regulation of organs is complex, involving multiple pathways that coordinates cell number (via the control of proliferation and apoptosis) and cell size. It is appreciated that mTORC1 works alongside the Hippo pathway to coordinate organ growth (reviewed in [98]). The transcription factor yes-associated protein 1 (YAP1) is activated in the Hippo pathway to drive gene-expression of CCND1 and Myc, which are involved in proliferation and cell size control, respectively. It is interesting that the protein translation of CCND1 and Myc mRNAs are mTORC1-dependent (Figure 1, and [65]). As a consequence, the Hippo and mTORC1 pathways would appear to work jointly to enhance organ growth by enhancing the transcriptional and translational regulation of CCND1 and Myc.
3.2. Energy Signalling and mTORC1
The anabolic processes needed for cellular growth are dependent on energy as well as nutrients, so mTORC1 enhances mitochondrial biogenesis to generate more mitochondria [99], ensuring that the cell has enough capacity to generate energy as the cell grows. The ability of mTORC1 to switch between states of anabolic growth and catabolic fasting is dynamically regulated and is intrinsically coupled with the energy senor, AMP-dependant protein kinase (AMPK). A major catabolic process that AMPK regulates is autophagy (reviewed in [100]). In the disease setting, improper mTORC1 signalling leads to uncontrolled cell growth and loss of energy homeostasis. Energy stress, through activation of AMPK is also upstream of TSC1/TSC2, where AMPK-dependent phosphorylation of TSC2 on Thr1227 and Ser1345 activates the TSC1/TSC2 tumour suppressor, leading to mTORC1 inhibition [101]. AMPK also phosphorylates Raptor, which is also inhibitory to mTORC1 [102]. AMPK becomes active when ATP levels decline and AMP levels increase. Upon energy stress, AMPK is activated by the serine/threonine kinase LKB1/STK11 [103,104]. Inactivating mutations of LKB1 inhibits AMPK and can give rise to Peutz-Jeghers syndrome that predisposes patients to hamartomatous polyps and cancer in the gastrointestinal tract [105]. As cells require a plentiful supply of energy to grow efficiently, this negative signalling input from AMPK ensures that cell growth through mTORC1 is halted when energy supply is insufficient. To quickly restore energy balance within the cell, AMPK not only downregulates anabolic processes driven by mTORC1, but also enhances catabolic processes, such as autophagy.
4. Conclusions
Since the discovery of rapamycin, much has been learnt about its drug target, mTORC1, and the role it plays in cell growth control. mTORC1 is regulated at the level of lysosomes, where multiple signalling inputs from growth factor (and hormone) receptors as well as from nutrients and energy all converge onto mTORC1. Coordination of such signalling inputs ensures that the homeostatic balance of nutrients and energy is maintained as a cell grows. The capacity of a cell to manufacture de novo proteins is rate limiting in cell growth, which is why the regulation of protein synthesis by mTORC1 is multifaceted through ribosomal biogenesis, generation of nucleotide precursors, and the regulation of protein translation. mTORC1 signalling is clearly tightly integrated with metabolism, but there is still much to understand. The nuclear function of mTORC1 is also currently unknown. Underdeveloped areas of research involving mTORC1 includes chromosomal remodelling and the regulation of transcription factors linked to cell growth control. mTORC1 is also implicated in mRNA splicing that adds another layer of regulation [106]. Clearly, there is still much to be discovered regarding how mTORC1 ‘mechanistically’ controls cell growth.
Acknowledgments
I would like to give special thanks to Health and Care Research Wales (the Wales Cancer Research Centre) and the Tuberous Sclerosis Association.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sehgal, S.N.; Baker, H.; Vézina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Park, S.R.; Yoo, Y.J.; Ban, Y.H.; Yoon, Y.J. Biosynthesis of rapamycin and its regulation: Past achievements and recent progress. J. Antibiot. 2010, 63, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, 7S–14S. [Google Scholar] [CrossRef]
- Schreiber, S.L. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 1991, 251, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Yanagida, M.; Maki, N.; Yagi, S.; Namiyama, F.; Kobayashi, T.; Hayano, T.; Takahashi, N.; Suzuki, M. Yeast cyclophilin-related gene encodes a nonessential second peptidyl-prolyl cis-trans isomerase associated with the secretory pathway. Transplant. Proc. 1991, 23, 2856–2861. [Google Scholar] [PubMed]
- Wiederrecht, G.; Brizuela, L.; Elliston, K.; Sigal, N.H.; Siekierka, J.J. FKB1 encodes a nonessential FK 506-binding protein in Saccharomyces cerevisiae and contains regions suggesting homology to the cyclophilins. Proc. Natl. Acad. Sci. USA 1991, 88, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Cafferkey, R.; Young, P.R.; McLaughlin, M.M.; Bergsma, D.J.; Koltin, Y.; Sathe, G.M.; Faucette, L.; Eng, W.K.; Johnson, R.K.; Livi, G.P. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol. Cell. Biol. 1993, 13, 6012–6023. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Stan, R.; McLaughlin, M.M.; Cafferkey, R.; Johnson, R.K.; Rosenberg, M.; Livi, G.P. Interaction between FKBP12-rapamycin and TOR involves a conserved serine residue. J. Biol. Chem. 1994, 269, 32027–32030. [Google Scholar] [PubMed]
- Lorenz, M.C.; Heitman, J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem. 1995, 270, 27531–27537. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Florentino, D.; Chen, J.; Crabtree, G.R.; Schreiber, S.L. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 1995, 82, 121–130. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Barbet, N.C.; Schneider, U.; Helliwell, S.B.; Stansfield, I.; Tuite, M.F.; Hall, M.N. TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 1996, 7, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, M.E.; Cutler, N.S.; Lorenz, M.C.; Di Como, C.J.; Heitman, J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 1999, 13, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.S.; Kuruvilla, F.G.; Tong, J.K.; Shamji, A.F.; Schreiber, S.L. Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 14866–14870. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Oppliger, W.; Hall, M.N. Molecular organization of target of rapamycin complex 2. J. Biol. Chem. 2005, 280, 30697–30704. [Google Scholar] [CrossRef] [PubMed]
- Bosotti, R.; Isacchi, A.; Sonnhammer, E.L. FAT: A novel domain in PIK-related kinases. Trends Biochem. Sci. 2000, 25, 225–257. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. Mammalian target of rapamycin complex 1: Signalling inputs, substrates and feedback mechanisms. Cell. Signal. 2009, 21, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Vander Haar, E.; Lee, S.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Takahashi, R.; Yoshino, K.; Tanimura, K.; Nakashima, A.; Eguchi, S.; Miyamoto, T.; Hara, K.; Takehana, K.; Avruch, J.; et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 2007, 282, 20329–20339. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. TORC2 Structure and Function. Trends Biochem. Sci. 2016, 41, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Bergman, P.; Triantafellow, E.; Wilson, C.J.; Zhu, Y.; Radetich, B.; Finan, P.M.; Klionsky, D.J.; Murphy, L.O. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol. Cell Biol. 2011, 31, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Stallock, J.P.; Ng, J.C.; Reinhard, C.; Neufeld, T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000, 14, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Huang, H.; Xu, T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 2001, 105, 357–368. [Google Scholar] [CrossRef]
- Gao, X.; Pan, D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001, 15, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Radimerski, T.; Montagne, J.; Hemmings-Mieszczak, M.; Thomas, G. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signalling. Genes Dev. 2002, 16, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Montagne, J.; Stewart, M.J.; Stocker, H.; Hafen, E.; Kozma, S.C.; Thomas, G. Drosophila S6 kinase: A regulator of cell size. Science 1999, 285, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Arrazola, P.; Hino, O.; Kobayashi, T.; Yeung, R.S.; Ru, B.; Pan, D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 2002, 4, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Kohrman, M.H. Emerging treatments in the management of tuberous sclerosis complex. Pediatr. Neurol. 2012, 46, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Sudentas, P.; Donohue, B.; Asrican, K.; Worku, A.; Walker, V.; Sun, Y.; Schmidt, K.; Albert, M.S.; El-Hashemite, N.; et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer 2005, 42, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.R.; Harris, P.C. The molecular genetics of tuberous sclerosis. Hum. Mol. Genet. 1994, 3, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Mach, K.E.; Furge, K.A.; Albright, C.F. Loss of Rhb1, a Rheb-related GTPase in fission yeast, causes growth arrest with a terminal phenotype similar to that caused by nitrogen starvation. Genetics 2000, 155, 611–622. [Google Scholar] [PubMed]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Tee, A.R.; Anjum, R.; Blenis, J. Inactivation of the tuberous sclerosis complex-1 and -2 gene products occurs by phosphoinositide 3-kinase/Akt-dependent and -independent phosphorylation of tuberin. J. Biol. Chem. 2003, 278, 37288–37296. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Beretta, L.; Gingras, A.C.; Svitkin, Y.V.; Hall, M.N.; Sonenberg, N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996, 15, 658–664. [Google Scholar] [PubMed]
- Raught, B.; Peiretti, F.; Gingras, A.C.; Livingstone, M.; Shahbazian, D.; Mayeur, G.L.; Polakiewicz, R.D.; Sonenberg, N.; Hershey, J.W. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004, 23, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Patel, H.R.; Takase, K.; Kohno, K.; Nairn, A.C.; Gelfand, E.W. Rapamycin selectively inhibits translation of mRNAs encoding elongation factors and ribosomal proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 11477–11481. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, H.B.; Fumagalli, S.; Dennis, P.B.; Reinhard, C.; Pearson, R.B.; Thomas, G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997, 16, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Crosio, C.; Boyl, P.P.; Loreni, F.; Pierandrei-Amaldi, P.; Amaldi, F. La protein has a positive effect on the translation of TOP mRNAs in vivo. Nucleic Acids Res. 2000, 28, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Zakaria, C.; Jia, J.J.; Graber, T.E.; Svitkin, Y.; Tahmasebi, S.; Healy, D.; Hoang, H.D.; Jensen, J.M.; Diao, I.T.; et al. La-related protein 1 (LARP1) represses terminal oligopyrimidine (TOP) mRNA translation downstream of mTOR complex 1 (mTORC1). J. Biol. Chem. 2015, 290, 15996–16020. [Google Scholar] [CrossRef] [PubMed]
- Lahr, R.M.; Fonseca, B.D.; Ciotti, G.E.; Al-Ashtal, H.A.; Jia, J.J.; Niklaus, M.R.; Blagden, S.P.; Alain, T.; Berman, A.J. La-related protein 1 (LARP1) binds the mRNA cap, blocking eIF4F assembly on TOP mRNAs. eLife 2017, 6, e24146. [Google Scholar] [CrossRef] [PubMed]
- Philippe, L.; Vasseur, J.J.; Debart, F.; Thoreen, C.C. La-related protein 1 (LARP1) repression of TOP mRNA translation is mediated through its cap-binding domain and controlled by an adjacent regulatory region. Nucleic Acids Res. 2018, 46, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Liu, H.; Zheng, X.F. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle 2010, 9, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Regulation of mammalian translation factors by nutrients. Eur. J. Biochem. 2002, 269, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Beugnet, A.; Tee, A.R.; Taylor, P.M.; Proud, C.G. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem. J. 2003, 372, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the Rag GTPases that signal amino acid to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A tumor suppressor complex with GAP activity for the RagGTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, U.S.; Karin, M. Sestrin regulation of TORC1: Is Sestrin a leucine sensor? Sci. Signal. 2016, 9, re5. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Ro, S.H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Layman, D.K.; Walker, D.A. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J. Nutr. 2006, 136, 319S–323S. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.M.; Tipton, K.D.; Aarsland, A.; Wolf, S.E.; Wolfe, R.R. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am. J. Physiol. 1997, 273, E99–E107. [Google Scholar] [CrossRef] [PubMed]
- Tumaneng, K.; Russell, R.C.; Guan, K.L. Organ size control by Hippo and TOR pathways. Curr. Biol. 2012, 22, R368–R379. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558.e18. [Google Scholar] [CrossRef] [PubMed]
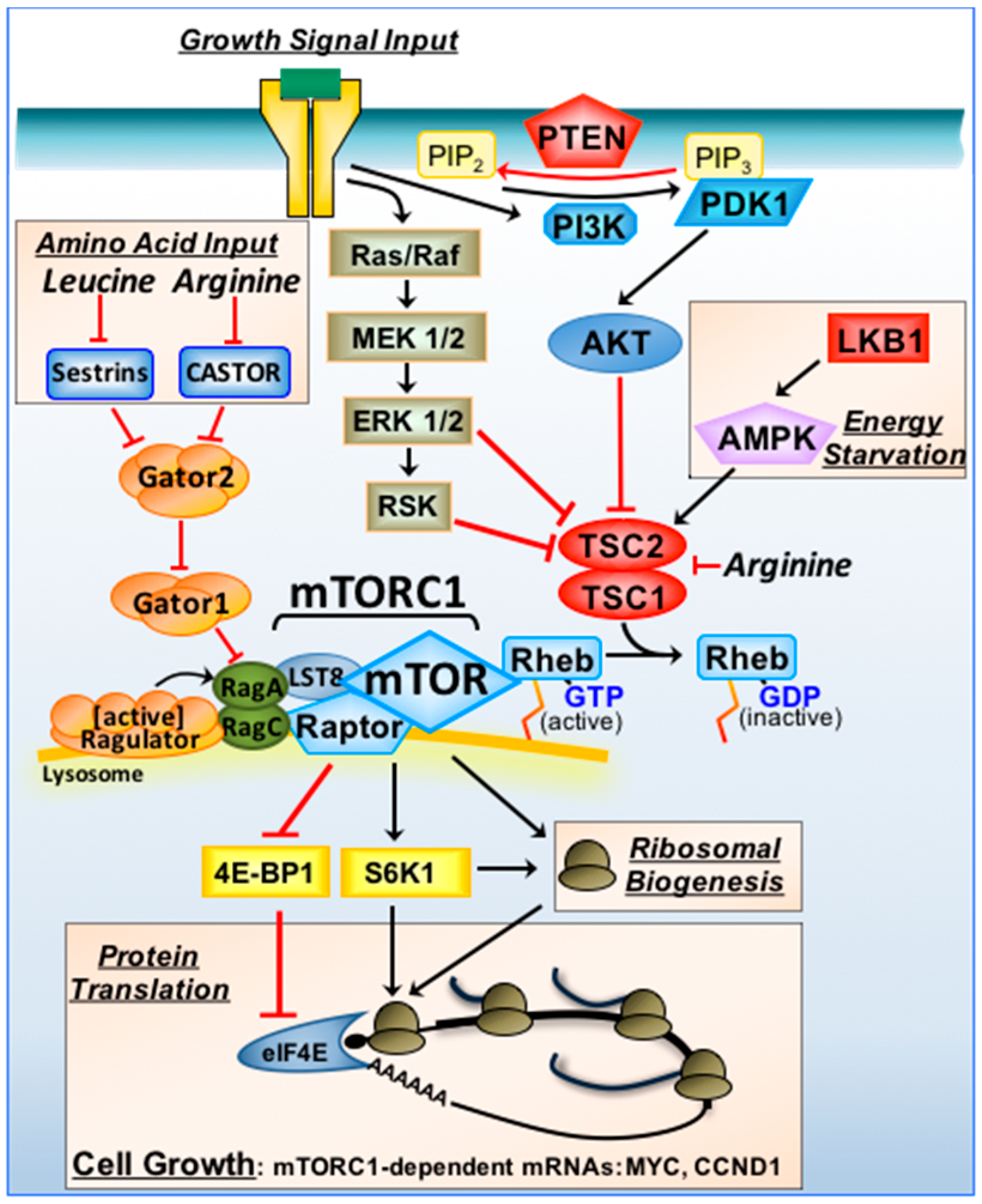
Figure 1.
mTORC1 signal transduction and tumour suppressors. Growth signals via plasma membrane bound receptors activate the Ras/Raf/MAPK/ERK/RSK and PI3K/AKT signalling pathways. Tumour suppressors upstream of TSC1/TSC2 include PTEN and LKB1 (indicated in red). Through these pathways, TSC1/TSC2 is inactivated, converting Rheb to an active GTP-bound state to promote mTORC1 (when associated with the “Ragulator complex” on lysosomal membranes). When nutrients are sufficient, Rag GTPase heterodimers recruit mTORC1 to the “Ragulator complex”. Arginine inhibits both TSC1/TSC2 and CASTOR. Leucine activates GATOR2 indirectly via sestrins. Under energy deprivation, LKB1/AMPK activates TSC1/TSC2 to switch mTORC1 off. mTORC1 drives cell growth (in part) by increasing the efficiency of mRNA translation of mTORC1-sensitive mRNAs (that include MYC and CCND1). mTORC1 regulates protein synthesis via 4E-BP1/eIF4E and S6K1.
Figure 1.
mTORC1 signal transduction and tumour suppressors. Growth signals via plasma membrane bound receptors activate the Ras/Raf/MAPK/ERK/RSK and PI3K/AKT signalling pathways. Tumour suppressors upstream of TSC1/TSC2 include PTEN and LKB1 (indicated in red). Through these pathways, TSC1/TSC2 is inactivated, converting Rheb to an active GTP-bound state to promote mTORC1 (when associated with the “Ragulator complex” on lysosomal membranes). When nutrients are sufficient, Rag GTPase heterodimers recruit mTORC1 to the “Ragulator complex”. Arginine inhibits both TSC1/TSC2 and CASTOR. Leucine activates GATOR2 indirectly via sestrins. Under energy deprivation, LKB1/AMPK activates TSC1/TSC2 to switch mTORC1 off. mTORC1 drives cell growth (in part) by increasing the efficiency of mRNA translation of mTORC1-sensitive mRNAs (that include MYC and CCND1). mTORC1 regulates protein synthesis via 4E-BP1/eIF4E and S6K1.

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tee, A.R. The Target of Rapamycin and Mechanisms of Cell Growth. Int. J. Mol. Sci. 2018, 19, 880. https://doi.org/10.3390/ijms19030880
AMA Style
Tee AR. The Target of Rapamycin and Mechanisms of Cell Growth. International Journal of Molecular Sciences. 2018; 19(3):880. https://doi.org/10.3390/ijms19030880
Chicago/Turabian StyleTee, Andrew R. 2018. "The Target of Rapamycin and Mechanisms of Cell Growth" International Journal of Molecular Sciences 19, no. 3: 880. https://doi.org/10.3390/ijms19030880
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.