PPAR Agonists and Metabolic Syndrome: An Established Role?
by
 and
and
Margherita Botta
1,†, 
Matteo Audano
1,†,
Amirhossein Sahebkar
2,3,4,
Cesare R. Sirtori
5,
Nico Mitro
1,* and
Massimiliano Ruscica
1
1
Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano, 20133 Milan, Italy
2
Biotechnology Research Center, Pharmaceutical Technology Institute, Mashhad University of Medical Sciences, Mashhad 9177948564, Iran
3
Neurogenic Inflammation Research Center, Mashhad University of Medical Sciences, Mashhad 9177948564, Iran
4
School of Pharmacy, Mashhad University of Medical Sciences, Mashhad 9177948564, Iran
5
Centro Dislipidemie, Azienda Socio Sanitaria Territoriale Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2018, 19(4), 1197; https://doi.org/10.3390/ijms19041197
Submission received: 27 March 2018
/
Revised: 10 April 2018
/
Accepted: 11 April 2018
/
Published: 14 April 2018
(This article belongs to the Special Issue PPARs in Cellular and Whole Body Energy Metabolism)
Abstract
:Therapeutic approaches to metabolic syndrome (MetS) are numerous and may target lipoproteins, blood pressure or anthropometric indices. Peroxisome proliferator-activated receptors (PPARs) are involved in the metabolic regulation of lipid and lipoprotein levels, i.e., triglycerides (TGs), blood glucose, and abdominal adiposity. PPARs may be classified into the α, β/δ and γ subtypes. The PPAR-α agonists, mainly fibrates (including newer molecules such as pemafibrate) and omega-3 fatty acids, are powerful TG-lowering agents. They mainly affect TG catabolism and, particularly with fibrates, raise the levels of high-density lipoprotein cholesterol (HDL-C). PPAR-γ agonists, mainly glitazones, show a smaller activity on TGs but are powerful glucose-lowering agents. Newer PPAR-α/δ agonists, e.g., elafibranor, have been designed to achieve single drugs with TG-lowering and HDL-C-raising effects, in addition to the insulin-sensitizing and antihyperglycemic effects of glitazones. They also hold promise for the treatment of non-alcoholic fatty liver disease (NAFLD) which is closely associated with the MetS. The PPAR system thus offers an important hope in the management of atherogenic dyslipidemias, although concerns regarding potential adverse events such as the rise of plasma creatinine, gallstone formation, drug–drug interactions (i.e., gemfibrozil) and myopathy should also be acknowledged.
1. Introduction
The incidence of metabolic syndrome (MetS), representing a global public health issue, has been estimated to vary from 20 to 27% in developing countries [1,2] to 35% in the USA [3]. MetS is a cluster of cardiometabolic risk factors, from high triglycerides (TGs), to elevated waist circumference (WC), high blood pressure (BP) and insulin resistance [4].
Following the first definition of MetS by the World Health Organization (WHO), several expert panels attempted to introduce stricter diagnostic criteria. In 2001, the National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) [5] recognized that the clustering of the metabolic risk factors included in the syndrome were indeed cardiovascular (CV) risk factors. In 2003, the American Association of Clinical Endocrinologists (AACE) modified the ATP III criteria highlighting the central role of insulin resistance in the pathogenesis of the syndrome [6]. In 2005, the International Diabetes Federation (IDF) issued a consensus document aimed at introducing a clinically useful definition of MetS in order to identify individuals at high risk of CV disease (CVD) and type 2 diabetes mellitus (T2D) on a worldwide basis [7]. In the same year, the American Heart Association (AHA)/National Heart, Lung and Blood Institute (NHLBI) suggested more specific criteria for the diagnosis of MetS [7]. Finally, a joint statement of IDF, NHLBI, AHA, World Heart Federation and International Association for the Study of Obesity, best known as the “Harmonization definition”, has been introduced and now represents the most commonly recognized criterion for the clinical diagnosis of MetS [4] (Table 1).
Carriers of MetS are at higher risk of developing atherosclerotic CVD, a condition worsened by the so called “atherogenic dyslipidemia”. This mixed dyslipidemia has emerged as the most clinically relevant “competitor” of elevated low-density lipoprotein cholesterol (LDL-C) among lipid risk factors. It is characterized by hypertriglyceridemia, low high-density lipoprotein (HDL)-cholesterol levels, and the prevalence of small, dense low-density lipoprotein (LDL) particles as well as an accumulation of cholesterol-rich remnant particles [9].
In this metabolic derangement, peroxisome proliferator-activated receptors (PPARs), i.e., nuclear receptors involved in the regulation of metabolic homeostasis, represent a valuable therapeutic target. PPAR activators have provided significant benefit in patients with primary hypertriglyceridemia (i.e., fibrates and omega-3 fatty acids, both PPAR-α agonists), as well as in cases of mixed hyperlipidemias with raised TGs and low HDL-C; conversely, PPAR-γ activators have become choice drugs in T2D.
PPAR agonists are generally recognized as effective pharmacological tools for the management of MetS [10,11]. A growing interest in PPAR activators has been acknowledged in recent years as they have been used in the more and more frequent occurrence of non-alcoholic fatty liver disease [12], the hepatic manifestation of MetS. However, post-marketing adverse effects should be recalled [13], i.e., weight gain, fluid retention, congestive heart failure, liver and gallbladder disease, renal effects, bone fractures, myopathy and rhabdomyolysis; these two last particularly with fibrates with an added risk when co-administered with statins [14,15].
Hence, the present review was aimed at discussing available evidence on new PPAR agonists in the clinical setting as well as at describing molecular mechanisms underlying the effects of these drugs. This review also attempts to reduce the current disagreement on the interpretation of outcomes of clinical trials with fibrates. To this end, we have revised and updated the available English-language studies relevant to the key clinical questions, published up to April 2018.
2. Peroxisome Proliferator-Activated Receptor: Key Players in Energy Homeostasis
PPARs are a subfamily of three ligand-inducible transcription factors, belonging to the superfamily of nuclear hormone receptors. In mammals, three different isoforms of PPARs have been described so far: PPAR-α, PPAR-β/δ and PPAR-γ. PPARs belong to the nuclear hormone receptor superfamily and, by binding to PPAR-responsive regulatory elements (PPRE), heterodimerise with the retinoid X receptor (RXR) and control a group of genes involved in adipogenesis, lipid metabolism, inflammation and maintenance of metabolic homeostasis [16,17,18,19,20]. PPAR-α is the first identified member and is mainly expressed in energy-demanding tissues that show high rates of β-oxidation (i.e., liver, kidney, heart and muscle). On the other hand, PPAR-β/δ is ubiquitously expressed in humans, whereas in mice it is expressed to a higher extent in the gastrointestinal duct, specifically stomach, large and small intestine. PPAR-γ is expressed at high levels in the adipose tissue.
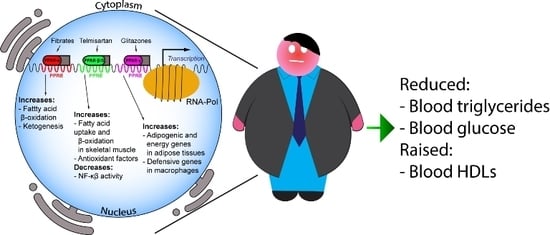
PPARs are activated by fatty acids and eicosanoids [21], as well as by small molecules, such as fibrates for PPAR-α, GW501516, GW0742, bezafibrate and Telmisartan for PPAR-β/δ, and glitazones for PPAR-γ. PPAR-α mediates the hypolipidemic function of fibrates in the treatment of hypertriglyceridemia and hypoalphalipoproteinemia [22], being the main regulator of intra- and extracellular lipid metabolism. Indeed, fibrates downregulate hepatic apolipoprotein C-III (ApoCIII) and stimulate lipoprotein lipase gene expression, thus being key players in TG metabolism [23].
Moreover, PPAR-α activation raises plasma HDL-C via induction of hepatic apolipoprotein A-I and apolipoprotein A-II expression in humans. On the other hand, glitazones exert hypotriglyceridemic activity by activating PPAR-γ, in turn inducing lipoprotein lipase expression in adipose tissue [23]. Finally, PPARs exert their function on intracellular lipid metabolism by regulating key proteins involved in the conversion of fatty acids to acyl-CoA esters, fatty acid import into mitochondria and peroxisomal and mitochondrial fatty acid oxidation [24,25]. Major roles and functions of the PPAR isotypes are depicted in Figure 1.
2.1. PPAR-α
PPAR-α (also called NR1C1) activation occurs mainly under energy deprivation. This leads to the upregulation of intracellular energy metabolism, ultimately inducing ATP production from oxidative phosphorylation. PPAR-α mRNA is upregulated in mouse liver during fasting, whereas PPAR-α knock-out (KO) fasted mice display significant hypoglycemia, hypoketonemia, hypothermia, and increased plasma free fatty acids, thus suggesting an inhibition of fatty acid uptake and oxidation [26]. PPAR-α-mediated fatty acid catabolism is crucial for the synthesis of several metabolites to be used as energy sources by other tissues such as ketone bodies in the brain [27]. Classical genes regulated by this nuclear receptor are the β-oxidative enzymes, e.g., carnitine palmitoyltransferase 1A and 2 (CPT1A and 2), acyl-CoA dehydrogenase very long chain (ACADVL), hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit-α (HADHA); similarly, important ketogenic genes like 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), 3-hydroxymethyl-3-methylglutaryl-CoA lyase (HMGCL) and acetyl-CoA acetyltransferase 1 (ACAT1) are stimulated by PPAR-α. This last seems to control liver glucose metabolism as well. Administration of fenofibrate to mice decreases expression levels of glucokinase and flux through this enzyme, suggesting a lower liver glucose uptake upon PPAR-α activation [28]. In another study, PPAR-α was found to induce pyruvate dehydrogenase kinase 4 (PDK4) expression, suppressing pyruvate transition to acetyl-CoA. Conversely, PPAR-α KO pups displayed a primary defect in gluconeogenesis, specifically from glycerol, leading to significant hypoglycemia [29].
PPAR-α induction also exerts an anti-inflammatory activity in mouse models, even though contrasting data are reported. The first evidence of PPAR-α involvement in the regulation of inflammation was provided by the group of Wahli more than 20 years ago [30]. The authors demonstrated that leukotriene B4 acts as a ligand for PPAR-α transcription, and the inflammatory response is prolonged in PPAR-α KO mice [31]. More recent studies have confirmed this association, showing that PPAR-α activation in mouse liver downregulates the CCAAT/enhancer binding protein β (C/EBPβ) as well as alpha (C/EBPα) and nuclear factor-κB (NFκB) protein expression, leading to lower levels of C-reactive protein, interleukin-6 and prostaglandins [32]. Conversely, dietary treatment with PPAR-α agonists increased lipopolysaccharide-induced plasma tumor necrosis factor α (TNF-α) levels that is instead reduced in PPAR-α-deficient mice, highlighting a possible role for PPAR-α also as a pro-inflammatory factor [33].
2.2. PPAR-β/δ
PPAR-β/δ (also called NR1C2) is the least well characterized isotype among PPARs. Nevertheless, it plays an important role in the metabolic adaptation of numerous tissues to environmental stimuli [34]. Physiologically, this isotype is activated by long-chain fatty acids, saturated and unsaturated, and by prostacyclin [35,36]. Notably, specific PPAR-β/δ activation leads to increased levels of fatty acid β-oxidation [37]. Moreover, PPAR-β/δ expression is upregulated specifically in skeletal muscle during fasting. These data were confirmed in PPAR-β/δ agonist treated L6 rat myocytes, showing increased fatty acid uptake and β-oxidation compared to controls [37].
Further studies suggest that PPAR-β/δ and physical exercise are tightly related; indeed, endurance training (6 weeks) boosted an up to 2.6-fold PPAR-β/δ protein expression in the tibialis anterior muscle [38]. The indispensable role of PPAR-β/δ in cell energy metabolism has been also shown in rat breast adenocarcinoma cells [39]. Specifically, the authors demonstrated that high PPAR-β/δ protein was associated with increased cancer cell growth in vitro and in vivo, suggesting that PPAR-β/δ favors breast cancer cell survival by regulating specific metabolic pathways.
PPAR-β/δ ligands have also been proposed as potential anti-inflammatory drugs [40,41]. Pharmacological activation of PPAR-β/δ in endothelial cells is associated with a potent anti-inflammatory effect, possibly by involving antioxidative genes and release of nuclear corepressors [42]. Moreover, investigation of PPAR-β/δ role in the modulation of NF-κB-driven inflammatory response confirmed the anti-inflammatory activity of this isotype [43]. The highly selective PPAR-β/δ agonist GW0742 in rats effectively antagonized lethality consequent to cecal ligation and puncture: drug treated animals had reduced release of pro-inflammatory cytokines and neutrophil infiltration in lung, liver, and cecum.
2.3. PPAR-γ
PPAR-γ (also called NR1C3) is mainly expressed in adipocytes (brown and white) and plays a major role in cell differentiation and energy metabolism [44,45]. Upregulation of PPAR-γ activity in vivo leads to bone loss and higher bone marrow adiposity, whereas downregulation leads to elevated bone mass [46]. The PPAR-γ agonists thiazolidinediones (TZDs) have a hypoglycemic action in ob/ob mice and improve insulin action in several models of obesity and diabetes [47,48]. A high correlation between the hypoglycemic activity of TZDs and their affinity for PPAR-γ has been repeatedly shown. While PPAR-γ-KO animals show embryonic lethality dying at 10.5–11.5 days postcoitum due to placental dysfunction, PPAR-γ heterozygote KO are characterized by higher insulin sensitivity and resistance to high-fat diet-induced insulin resistance [49,50,51].
PPAR-γ Pro12Ala partial loss-of-function mutation in humans leads to decreased body mass index, higher insulin sensitivity and protection from T2D [52]. The most accepted hypothesis is that PPAR-γ facilitates energy storage after high-fat diet challenge, partly by suppressing leptin expression in adipocytes [49]. In addition, PPAR-γ directly binds the promoter of almost all adipogenic genes, such as factors involved in glucose and fatty acid metabolism, indicating that this isotype is fundamental for the activation of metabolic programs during adipogenesis [53]. While PPAR-γ is not needed for macrophage differentiation, it is fundamental for a proper anti-inflammatory activity in adipose tissue macrophages [54]. This specific feature of PPAR-γ is due to cell type specific DNA binding, as shown in macrophages, where PPAR-γ cooperates mainly with the spleen focus forming virus (SFFV) proviral integration into the oncogene transcription factor [55]. On the other hand, it is well established that CCAAT/enhancer binding protein (C/EBP) α and β transcription factors are major partners of PPAR-γ during adipocyte differentiation [53].
Comparative analysis of PPAR-γ binding patterns in adipocytes and macrophages indicates that it binds to immune defense genes in macrophages, whereas the only cluster of genes shared with adipocytes is the one encoding metabolic factors [55]. Notably, myeloid lineage-specific PPAR-γ KO mice display lower alternative macrophage activation and a strong metabolic phenotype, characterized by diet-induced obesity, insulin resistance, and glucose intolerance [55]. In addition, PPAR-γ activation decreases T-lymphocyte-dependent inflammation of adipose tissue and development of insulin resistance in diet-induced obese mice [56]. Recently, it has been demonstrated that the PPAR-γ agonist pioglitazone may prevent or delay aortic aneurysm progression in patients [57]. Treated patients show decreased macrophage infiltration into the retroperitoneal periaortic fat, as well as tumor necrosis factor α (TNFα) and matrix metallopeptidase 9 (MMP9) gene expression. On the other hand, treatment increased adiponectin expressions in both tissues compared to controls. Neurons are protected by pioglitazone treatment [58]; indeed, neuron and axon susceptibility to both nitric-oxide donor-induced and microglia-derived nitric oxide-induced toxicity are reduced, whereas catalase expression is raised [58].
3. PPAR-α: Fibrates and Omega-3 Fatty Acids in the Metabolic Syndrome
The characteristic features of MetS, i.e., increased triglyceridemia, abdominal obesity, reduced HDL-C levels and increased glycemia, in addition to raised blood pressure, clearly indicate that PPAR-α agonists have an ideal profile to control most of these features [59].
3.1. Fibrates
The role of fibrates in the clinical management of disorders characterized by elevated TGs is now well established [60]. Fibrates, activators of the PPAR system, mainly PPAR-α, have shown significant benefit in clinical trials of CV prevention, i.e., reducing the occurrence of nonfatal myocardial infarction, particularly when restricting evaluation to patients with concomitant TG elevation and HDL-C reduction [61].
Clinical findings, at times disappointing (FIELD—Fenofibrate Intervention and Event Lowering in Diabetes—study) [62], have been hampered by inappropriate patient selection and, possibly, by the use of non-optimal drug formulations [63,64]. On the other hand, long-term re-evaluation of most recent studies, particularly the ACCORD—Action to Control Cardiovascular Risk in Diabetes—trial with fenofibrate, has clearly indicated that the agent has a heterogeneous response, but may have a valid indication in reducing CVD in appropriately selected patients, i.e., those with diabetes, hypertriglyceridemia and low HDL-C [65]. In these subjects, a definite benefit on the CV outcomes may be predicted.
When projected to the growing field of lipid-lowering treatments in clinical practice, these observations have fostered the development of newer pharmacological approaches, partially based on the PPAR target, but providing additional or alternative mechanisms which may lead to lipoprotein changes and altered glycemic parameters, not found with the established agents. Hence, this pharmacological approach may be of potential value in the clinical approach to the more and more frequent occurrence of MetS [66].
A recent meta-analysis from 22 RCTs involving a total of 11,402 subjects showed that fibrate administration decreased fasting plasma glucose (−5 mg/dL), insulin levels (−0.56 µIU/mL), and insulin resistance (HOMA-IR: −1.09), but not HbA1C. This latter evidence may be due to the short-term duration—less than three months—of the included studies. Moreover, the reduction in glucose, although statistically significant, was small and, thus, not clinically relevant [67]. These findings have been well characterized from studies with fenofibrate and bezafibrate. This latter, differently from the most selective PPAR-α modulator fenofibrate, acts as a pan PPAR activator for all three PPAR isoforms (α, γ and δ) [68]. Bezafibrate, similar to the selective PPAR-γ agonists, i.e., thiazolidinedione (TZDs) that are specifically used in T2D treatment, may exert a glucose-lowering activity but apparently without causing water retention, weight gain and peripheral edema that are potential side effects of glitazones [69,70]. Bezafibrate, compared with other fibrates, reduces the incidence of T2D, with a better reduction of blood glucose, HbA1C and insulin resistance [71].
Fibrates: Evidence from the Most Recent Clinical Trials
Pemafibrate (formerly known as K-877) is one of the newest members of the selective PPAR-α modulators, being >2000-fold more selective for PPAR-α vs. either PPAR-γ or -δ (delta) [72]. Pemafibrate has been recently approved in Japan for the treatment of hyperlipidemias, with a recommended dosage of 0.1 mg bid with the possibility of reaching a maximum of 0.2 mg bid [73].
The long-term efficacy of pemafibrate has been recently reported in a phase 3 multicenter, placebo-controlled, randomized, double-blind, parallel-group study (JapicCTI-142412) on T2D patients (HbA1c ≥ 6.2%) with fasting TGs ≥ 150 mg/dL, not on statins. The primary endpoint was the percentage change in fasting TG levels from baseline and secondary endpoints were changes in fasting and postprandial lipoproteins and glycemic parameters. Among the 167 eligible participants (mean age 60.5 years), 54 and 55 were randomized to pemafibrate 0.1 or 0.2 mg bid, respectively, and 57 to a placebo. Twenty-four weeks of treatment led to a significant 45% decrement of TGs regardless of dose; fasting TGs ≤ 150 mg/dL were achieved by 81.5% and 70.9% of patients on 0.2 and 0.4 mg/day, respectively; statistically significant when compared to the placebo group. In addition, non-HDL-C, cholesterol remnants, ApoB100, ApoB48, and ApoCIII levels were reduced with a concomitant rise of HDL-C and ApoA-I (Table 2) [74]. The reduction of ApoCIII levels confirmed the general activity of PPAR activators on this variable, in line with what reported with statin treatments [75].
Notably, pemafibrate led to a more anti-atherogenic profile, i.e., higher levels of medium, small and very small HDL particles vs very large and large particles at baseline. Although no changes were seen in LDL-C levels, a significant increment of large LDL and a reduction of small and very small particles were found in the pemafibrate arms. Modest changes were seen in glycemic parameters: only the 0.2 mg dose significantly reduced the HOMA-insulin resistance score with no significant changes in fasting glucose, insulin, glycated albumin and HbA1c. Both pemafibrate doses significantly raised circulating levels of FGF-21 [74]. All groups displayed comparable rates of adverse events and drug reactions, i.e., serum creatinine and liver enzyme increases.
The efficacy of pemafibrate over that of fenofibrate was reported in a 24-week, randomized, double blind, active-controlled, phase 3 trial. Patients with fasting TG ≥ 150 mg/dL as well as HDL-C ≤50 mg/dL for men and ≤55 mg/dL for women were randomly assigned to pemafibrate 0.1 (n = 73) or 0.2 (n = 74) mg bid or to fenofibrate 106.6 mg qd (n = 76). The primary efficacy analysis, i.e., percent change in fasting TG from baseline, demonstrated that pemafibrate treatments reduced TGs around −46% vs. −39.7% for fenofibrate. These findings can be translated into a further −6.5% and −6.2% difference in TG reduction for patients on 0.2 and 0.4 mg/day pemafibrate vs. fenofibrate. TC, non-HDL-C, ApoB, and VLDL-C were significantly decreased, and HDL-C, ApoAI, and ApoAII increased by both agents with no significant differences among treatment groups. Conversely, FGF-21 levels were raised to a greater extent in the 0.4 mg/day pemafibrate group vs. fenofibrate [76] (Table 2). Adverse drug reactions, such as rises in liver enzymes and serum creatinine, were observed in the fenofibrate group, not in the pemafibrate groups.
The non-inferiority of pemafibrate over fenofibrate was confirmed in a 12-week phase 3 trial enrolling 489 patients with TG ≥ 200 mg/dL and HDL-C ≤ 50 mg/dL. The TG lowering effects of pemafibrate were dose dependent −46.3% (0.1 mg/day), −46.7% (0.2 mg/day) and −51.8% (0.4 mg/day) and non-inferior to those of fenofibrate, −38.3% (100 mg/day) and −51.5% (200 mg/day) (Table 2). Adverse events were less frequent than with fenofibrate 200 mg/day [77].
The long-term efficacy of pemafibrate to treat residual hypertriglyceridaemia during statin treatment has been recently evaluated in two randomized, double-blind, placebo-controlled phase 2 trials. The primary endpoint was the percentage changes in fasting TGs from baseline [78]. The first trial study enrolled 188 patients with residual dyslipidemia (fasting TGs from 347 to 382 mg/dL) on a pitavastatin background with LDL-C in the range of 116–125 mg/dL. The 12-week pemafibrate administration (0.1, 0.2 or 0.4 mg/day) significantly reduced TG levels by −46.1%, −53.4% and −52%, respectively. Conversely, no TG reduction was observed in the pitavastatin monotherapy group. Combination therapy led to a significant rise of HDL-C (range: +12.7–19.7%), ApoAI (range: +1.5–6.6%) and ApoAII (range: +18.5–27.6%) and a reduction of non-HDL-C (range: −10.7–13.1%) and ApoB (range: −7.9–8.6%) (Table 2). Notably, all of these changes were statistically significant when compared to pitavastatin alone. Pemafibrate as an add-on therapy resulted in a more anti-atherogenic lipoprotein profile, i.e., increment of cholesterol in medium, small, and very small HDL subclasses and in large and medium LDL subclasses [79].
In the second trial, pemafibrate (0.2 mg/day) was given for 24 weeks to 423 patients with residual dyslipidemia (TGs ranging from 325 to 333 mg/dL) on statins (most commonly atorvastatin, rosuvastatin and pitavastatin); LDL-C was around 108 mg/dL. Notably, if TGs were ≥ 150 mg/dL after 12 weeks, pemafibrate was up-titrated to 0.4 mg/dL. Regardless of statin background, combination therapy with pemafibrate 0.2 or 0.4 mg/day led to TG reductions of about 50% from baseline [79] (Table 2). Compared to the monotherapy arm, patients receiving pemafibrate showed a significant decrement in non-HDL, ApoB and ApoCIII, as well as an increment in HDL-C, ApoAI and ApoAII [79]. As previously described, the addition of pemafibrate led to an increment in medium and small lipid-poor HDL, more efficient in reverse cholesterol efflux [80]. In both of these last two studies, the incidence of adverse events during treatment was similar across all groups. The proportion of patients experiencing elevated alanine transaminase (ALT), creatine kinase (CK) and serum creatinine were comparable.
The clear definition of efficacy of pemafibrate on the lipid profile, i.e., TG reduction and HDL-C increment, in preclinical studies as well as in phase 1 and phase 2 clinical trials (previously reviewed [73,81]) led to the planning of the PROMINENT (pemafibrate to reduce cardiovascular outcomes by reducing triglycerides in patients with diabetes) trial (registered as NCT03071692). The primary objective of this phase 3 study is evaluation in T2D patients already on statin (fasting TGs: ≥200 to <500 mg/dL; HDL-C ≤ 40 mg/dL), testing whether pemafibrate (0.2 mg bid) can delay the time of the first occurrence of nonfatal myocardial infarction (MI), nonfatal ischemic stroke, hospitalization for unstable angina requiring unplanned coronary revascularization, and CV death. Changes in lipid end-points including ApoAI, ApoCIII, ApoE and non-fasting remnant cholesterol are listed as secondary outcomes [82].
The efficacy of fibrates on CV prevention has been disputed, mainly on the ground of studies on non-selected patients [63,64]. Long-term re-evaluation of some of the most recent trials has clearly confirmed that fenofibrate in particular may have a valid indication in reducing CVD in patients with diabetes, hypertriglyceridemia and low HDL-C [65]. The recently available low-dosage pemafibrate seems to provide a higher activity on the HDL system and the ongoing studies on vascular prevention will provide further data on the link between biochemical markers of the MetS and CV risk.
3.2. Omega-3
Fatty acids of the n-3 series (i.e., with multiple double bonds, the first one being in the n-3 position from the terminal methyl group) have provided an important addition to the dietary treatment in syndromes characterized by elevated TGs. Omega-3s act as “fraudulent fatty acids” [83] i.e., they, somewhat similar to drugs with a fatty acid-like structure, particularly fibrates, do not enter the liver metabolic handling by the classical fatty acetylCoA oxidative mechanism, with carnitine mediated transport to mitochondria [84]. They are instead handled by a non-mitochondrial regulated pathway, differently expressed both in the liver and other tissues [85]. Peroxisomal associated receptors are stimulated in the presence of those fatty acids catabolized not only by the classical mitochondrial pathway [86]. Both fibrates and omega-3s, thus, will not act as classical substrates of mitochondrial metabolism, but rather stimulate the metabolism of fatty acids coming from diet or end products of, e.g., TG metabolism by the PPAR-α mediated pathway. Peroxisomal stimulation is less extensive than in the case of fibrates, but it can well stimulate fatty acid oxidation. An additional mechanism, more closely related to the plasma glucose elevation in MetS, is the activation of tissue glucose uptake by the GLUT4 transporter in adipocytes; this mechanism appears to be mediated by the GPR120 protein, functioning as an omega-3 fatty acid receptor/sensor [87].
Controlled trials in patients given relatively elevated daily doses of omega-3 in the form of TGs or, more recently, of ethyl esters of eicosapentaenoic (EPA) and docosahexaeneoic (DHA) acids, as well as with novel formulations of separate fatty acids [88], have repeatedly confirmed a TG reduction in hypertriglyceridemic conditions associated or not with diabetes [89,90]. Controlled studies indicate lowering, in general, of 20–30% of fasting triglyceridemia in these conditions [91], with a moderate rise of HDL cholesterolemia as well as an increment in reverse cholesterol transport mainly by influencing HDL remodeling and promoting hepatobiliary sterol excretion [92].
A general review on the mechanism of omega-3, improving abnormalities characteristic of MetS, may in addition to the classical activation of fatty acid metabolism involve increased adipocyte differentiation, reduced lipolysis and lipogenesis, as well as a significant activity on low grade inflammation, including reduced adipokines and specialized pro-resolving lipid mediators [93]. By the activation of fat metabolism and consequent energy expenditure, it being peroxisomal and to some extent also mitochondrial, positive effects on obesity may be observed. Indeed, PPAR-α KO-obese mice show, in fact, a clear worsening of obesity that may be instead improved by omega-3 administration in different diet-induced conditions [94]. Reduced lipogenesis and low-grade inflammation may be of value in the treatment of complex metabolic disorders.
In the case of adipose tissue biology, the most recent evidence points out to the importance of both white and brown adipose tissue (WAT and BAT) function. “Healthy adipocytes” in WAT are relatively small fat cells with a high capacity for mitochondrial oxidative phosphorylation, TG/FA cycling and de novo lipogenesis [94]. These cells, with a flexible phenotype, may provide beneficial local and systemic effects by protecting against inflammatory responses during lipolysis, preventing fat accumulation and dyslipidemia caused by increased liver VLDL-TG synthesis.
Indeed, dietary omega-3s may indeed redirect adipose tissue to a “healthy phenotype” [94]. These polyunsaturated fatty acids appear to stimulate the “G protein coupled receptor” GPR120 [95], promoting BAT activation [96], thus inducing brown as well as beige adipocyte differentiation and thermogenic activation which seems to be linked to an increase in blood FGF-21 levels. Characteristically, in animals devoid of GPR120, adipose tissue thermogenic activation is not achieved [97]. These observations are of ethnological significance, in view of the high consumption of omega-3 from fish in individuals living in cold areas such as the Eskimos [98]. Interestingly, in in vitro systems only BAT and not WAT cells synthesize DHA [99].
Inflammatory changes in the adipose tissue are characteristic of obesity. They are driven by rises in circulating endotoxins and infiltrate immune cell populations. This will lead to an increased secretion of inflammatory adipokines (e.g., IL-6, TNF–α, monocyte chemoattractant protein (MCP) and chemokine (C-C motif) ligand (CCL) from multiple cellular sources [100]. The end result of the increased secretion of inflammatory mediators is the development of insulin resistance. A characteristic reduction in chemokine secretion from LPS stimulated co-cultures of omega-3 fed rodents is followed by a reduced secretion of IL-6 (−42%) and TNF-α (−67%), as well as by a similar reduction of other inflammatory mediators. Concomitantly, omega-3s increase the mRNA expression of negative regulators of inflammatory signaling, such as the monocyte chemoattractant 1-induced protein (MCPIP; +9.3-fold) and the suppressor of cytokine signaling 3 (SOCS3; +1.7-fold) [101]. In patients with MetS a reduction of high sensitivity CRP levels [102] has been found, although data from a number of clinical studies on this topic have been inconsistent [91,103].
It can thus be concluded that, in the adipose tissue, besides the stimulation of fatty acid catabolism, well-characterized anti-inflammatory and anti-chemotactic effects can be exerted by omega-3s. All these effects, recognized at the cellular level, may be followed by biochemical changes in patients with MetS and diabetes mellitus. In these patients, statistically significant TG reductions, compared to placebo, have been observed with, however, somewhat differential effects on LDL-C. Apparently, products containing DHA may increase LDL-C levels, whereas those containing EPA only products do not lead to a similar consequence [104,105].
Omega-3: Evidence from the Most Recent Clinical Trials
Most recently, the effect of omega-3 fatty acids (2 g daily) in reducing TGs and other lipid concentrations in patients with severe hypertriglyceridemia (TG > 500 mg/dL and <2500 mg/dL) was evaluated in the EVOLVEII (Epanova® for lowering very high triglycerides II) trial, a double-blind, randomized, olive oil-controlled study. After an 8-week screening period for patients who required washout or stabilization of lipid-lowering therapy (e.g., statin or cholesterol-absorption inhibitors), they were randomized to receive two 1 g soft gelatin capsules with omega-3 (550 mg EPA + approximately 200 mg DHA in a new formulation) or olive oil once a day for 12 weeks. Notably, stratification was carried out based on TG levels, i.e., ≥ 500 ≤ 885 mg/dL or > 885 < 2500 mg/dL. Omega-3 capsules reduced TG by −28.1% vs. −10.2% (olive oil) in the group with TGs ≥ 500 and by −37.5% vs. −9.3% (olive oil) in the group with TGs > 885 mg/dL. In the whole population, TG differences between the two treatment groups were −14.2%. Non-HDL-C percentage changes were instead −8.8% (omega-3) vs. +0.4% (olive oil) in the group with TGs ≥ 500, with more marked differences in those with TG > 885 (−14% vs. +3.1%), and an overall −9% non-HDL-C reduction (Table 2). Omega-3 supplementation led to a significant lowering of VLDL-C, both when compared to baseline or to the olive oil arm. The decrease of VLDL-C concentrations was similar to that of TGs. HDL-C were modestly raised by both treatments, with no extra benefit given by omega-3 [106].
The results of this trial are in line with those found in the previous EVOLVE (Epanova for lowering very high triglycerides) double-blind, randomized, parallel, 4-arm study. In subjects with severe hypertriglyceridemia (TGs ≥ 500 mg/dL but <2000 mg/dL), administration of omega-3-FA 2 g/die (plus olive oil 2 g/day), omega-3FA 3 g/die (plus olive oil 1 g/day), or omega-3-FA 4 g/day for 12 weeks in combination with diet and lifestyle changes led to a −31% reduction in fasting TG in the group receiving omega-3-FFA 4 g/die vs. 25% in the other two treatment groups. A minimal TG reduction (−4.3%) was found in patients receiving olive oil 4g/day. A similar trend was found for non-HDL-C, with a maximal −9.6% reduction with omega-3-FA 4 g/day, vs. a +2.5% increment in the olive oil group (Table 2). HDL-C were not significantly changed at any dosage [107].
The effect of omega-3 as an add-on therapy to a statin background was evaluated in the ESPRIT (Epanova combined with a statin in patients with hypertriglyceridemia to reduce non-HDL cholesterol) trial, on persistently hypertriglyceridemic patients already on a maximally tolerated dose of statin or statin + ezetimibe, with TG levels ≥ 200 mg/dL and <500 mg/dL). Compared to olive oil (4 g/day), omega-3-FA 2 g/day or omega-3-FA 4 g/day administration led to a significant reduction in non-HDL-C (−3.9% and −6.9%, respectively) and TG (−14.6% and −20.6%, respectively) (Table 2) [108].
A limited number of studies have selectively evaluated the efficacy of omega-3 fatty acids on CV outcomes, both in primary and secondary prevention. Positive outcomes were reported from the GISSI (Gruppo Italiano per lo studio della sopravvivenza nell’infarto miocardico) [109] study which dealt mainly with patients with an acute coronary syndrome, and from the JELIS (Japan eicosapentaenoic acid lipid intervention study) [110] testing the efficacy of omega-3 (1800 mg/day) in primary prevention moderately hypercholesterolemic patients, mainly on statins. In contrast, the large alpha omega trial on coronary patients on a smaller daily intake of EPA + DHA (400 mg/day) failed to reach the targeted reduction of CV events [111]. While these last authors, in a recent meta-analysis on 77,917 patients in 10 different studies, appeared to confirm the lack of a significant impact of omega-3 on CV endpoints [112], at present, the effect of omega-3 supplementation on CV outcomes, i.e., any component of the composite of major adverse cardiac events (MACE), is being evaluated in the STRENGTH (study to assess statin residual risk reduction with Epanova in high cardiovascular risk patients with hypertriglyceridemia) and REDUCE-IT (reduction of cardiovascular events with icosapent ethyl–intervention) trials [113] targeting particularly hypertriglyceridemic patients.
Unquestionably, the efficacy of omega-3 intake on metabolic parameters and on inflammatory changes is now well established, with a convincing series of mechanistic studies, but the efficacy of these nutritional supplements on CV outcomes is at present unsettled. The ongoing studies should shed light on this last issue, particularly as pertains to patients with hypertriglyceridemia associated risk.
4. PPAR-γ Agonists
Agonists of PPAR-γ belong to the thiazolidinedione (TZDs) drug class and are currently in use for T2D [114]. Pioglitazone and rosiglitazone are the only two drugs available. Following epidemiological data indicative of a raised CV risk after rosiglitazone [115,116], the drug was taken off the market in Europe. Pioglitazone appears instead to reduce CV events [117]. Clinical trials with pioglitazone, e.g., PERISCOPE, PROactive and CHICAGO, have, in fact, demonstrated that in addition to beneficial effects in reducing TGs and increasing HDL-C levels, pioglitazone can reduce CV risk in T2D patients [118].
The PERISCOPE trial (pioglitazone effect on regression of intravascular sonographic coronary obstruction prospective evaluation) recruited patients with baseline HbA1c ≥ 6.0% to ≤9.0% (if on a glucose-lowering medication) or ≥6.5% to ≤10% (if not on drug therapy) with positive coronary angiogram (at least 1 angiographic stenosis with at least 20% narrowing). Pioglitazone, compared with glimepiride, raised HDL-C by +5.7 mg/dL vs. 0.9 mg/dL, whereas TG levels were decreased by −16.3 mg/dL vs. a rise of +3.3mg/dL. Fasting insulin levels were decreased by pioglitazone and raised by glimepiride. The primary end-point, namely the percent atheroma volume change measured by intravascular ultrasound (IVUS), was reduced by −0.16% in the pioglitazone arm vs. a +0.73% rise in the glimepiride group (Table 2); these between group changes were statistically significant [119]. Interestingly, a post-hoc analysis showed a greater relative increase in HDL-C (+14.2% vs. 7.8%) with a concomitant relative reduction of TGs (−13.3% vs. −1.9%), TG/HDL-C ratio (−22.5% vs. −9.9%) and HbA1C (−0.6% vs. −0.3%) upon pioglitazone administration, possibly responsible for the atheroma regression [120].
The CHICAGO (carotid intima-media thickness in atherosclerosis using pioglitazone) trial tested the hypothesis that pioglitazone would have a beneficial effect in reducing carotid intima-media thickness (IMT) progression compared with glimepiride [121]. The reported reduced progression of IMT appeared to be associated with a rise of HDL-C (+14%) following pioglitazone (Table 2) [122]. The positive effect of pioglitazone on carotid IMT is thus independent of its glucose lowering effect. Of note, weight and body mass index were higher in the pioglitazone arm.
The IRIS (insulin resistance intervention after stroke) trial showed that pioglitazone reduced the occurrence of fatal and non-fatal stroke or MI in insulin resistant patients without diabetes, also halving the occurrence of diabetes (Table 2). Notably, this latter occurred in 3.8% of patients on pioglitazone vs. 7.7% of those assigned to placebo (hazard ratio: 0.48; 95% CI: 0.33–0.69) [123,124]. Evaluation of safety outcomes indicated that pioglitazone led to (i) a weight gain of 4.5 kg (52.2% of patients) vs. +13.6 kg (11.4% of patients) for placebo [123]; with (ii) an increment in the absolute risk of fractures risk by 1.6% vs. 4.9%, depending on fracture classification [125]; and (iii) a higher incidence of edema (+35.6% vs. +24.9%) [123], this last being an as yet poorly understood frequent side effect of glitazones [126].
The clinical outcome studies on PPAR-γ agonists have been focused mainly on pioglitazone, particularly in view of the better tolerability. These have concluded that this treatment may be associated both with a reduced atheroma progression and a lower incidence of diabetes. Further, the IRIS study provided clear evidence of the efficacy of pioglitazone in preventing CV outcomes in insulin resistant patients [123,124].
5. PPAR Dual Agonists
Dual PPAR agonists or partial agonists, e.g., dual α/γ, α/δ or β/δ [127] were developed with the aim of achieving the TG-lowering and HDL-raising effects of PPAR-α activators as well as the insulin-sensitizing and antihyperglycemic effects of TZDs with a single drug. Such a combination of effects would be ideal for the treatment of T2D, MetS and NAFLD, which all share as common features atherogenic dyslipidemia and insulin resistance [128].
Of particular interest is the case of NAFLD, for which dual PPAR-α/δ agonists offer significant hope. Elafibranor (formerly known as GFT-505), with preferential α (EC50 = 6 nM) and complementary δ (EC50 = 47 nM) receptor agonist activity, is targeted to the liver, where it is converted to the main active metabolite, GFT-1007, in a dose-dependent manner. Elafibranor has been shown to be effective in disease models of NAFLD/NASH and liver fibrosis [129], as well as in T2D patients for whom a reduction in TG and LDL-C levels and improved insulin sensitivity were reported [130]. Recent results from the Phase 2b GOLDEN trial (NCT01694849) indicated that elafibranor treatment leads to a substantial histological improvement of NASH, including resolution of steatohepatitis and reduced CV risk. NASH was resolved without fibrosis worsening in 23% and 21% of patients assigned to receive either 80 mg or 120 mg/day elafibranor vs. 17% in the placebo arm; no significant differences between groups were found. When a more stringent definition of NASH was considered, changes in NASH resolution were 19% after elafibranor administration vs. 12% in the placebo group (p = 0.045) (Table 2) [131].
For activators of PPAR α + γ receptor, only preliminary data are available for saroglitazar, i.e., positive effects on the lipid profile, blood pressure, atherosclerosis, inflammation, and clotting [132]. Saroglitazar is being tested in an ongoing phase 3 trial in non-cirrhotic biopsy-proven NASH patients, in order to evaluate a possible improvement in NASH histology without worsening of fibrosis [133]. Interestingly, this agent is currently approved in India for the treatment of diabetic dyslipidemia [134]. Other glitazars, i.e., tesaglitazar and rasaglitazar have been discontinued from clinical development due to renal side effects, anaemia and leukopenia (tesaglitazar) and bladder tumor development (ragaglitazar) [135].
At present, dual agonists show an attractive metabolic profile, including an activity on MetS associated liver abnormalities. Clinical outcome studies are awaited with interest.
6. Conclusions
The management of MetS represents one of the major targets in atherosclerosis prevention. While treatment of hypercholesterolemia or diabetes can be successfully handled with drugs targeting cholesterol biosynthesis or beta-islet-cell function, MetS is characterized by a number of diverse metabolic abnormalities which are more difficult to pursue. While lifestyle modifications [136], including changes in diet and increased exercise, can provide help to a small number of patients, an improved knowledge of drugs affecting PPAR system has led to more frequent and better focused treatment choices. PPAR agonist “fraudulent fatty acids”, i.e., fibrates and omega-3 fatty acids, find a growing role in the handling of hypertriglyceridemia and, in the case of fibrates, also positively affecting HDL-cholesterol and the consequently raised CV risk. PPAR-γ agonists are instead targeted to the glycemic abnormalities of MetS; they may, however, lead to weight increase. The causal role of hypertriglyceridemia as a CV risk factor, confirmed by Mendelian randomization studies, has brought clinicians back to this somewhat forgotten risk marker, now rated by many as an unmet need [137]. The HDL-C raising approach has also become of high interest after the preventive failure of drugs such as the cholesteryl ester transfer protein inhibitors [138], disclosing the as yet not fully clarified CV protective mechanism of HDL [139].
Acknowledgments
This work was supported by Fondazione Cariplo grant No. 2015-0552; Linea 2, Azione (Intramural grant) received by Università degli Studi di Milano and by the Fondazione Carlo Sirtori.
Author Contributions
Margherita Botta and Matteo Audano wrote the manuscript; Amirhossein Sahebkar and Cesare R. Sirtori critically revised the text; Nico Mitro and Massimiliano Ruscica specifically examined the molecular aspects of PPAR activation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Carvalho Vidigal, F.; Bressan, J.; Babio, N.; Salas-Salvado, J. Prevalence of metabolic syndrome in Brazilian adults: A systematic review. BMC Public Health 2013, 13, 1198. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, W.; Lun, Z.; Zhang, H.; Sun, Z.; Kanu, J.S.; Qiu, S.; Cheng, Y.; Liu, Y. Prevalence of metabolic syndrome in Mainland China: A meta-analysis of published studies. BMC Public Health 2016, 16, 296. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed]
- National Cholesterol Education Program (NCEP); Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- Einhorn, D.; Reaven, G.M.; Cobin, R.H.; Ford, E.; Ganda, O.P.; Handelsman, Y.; Hellman, R.; Jellinger, P.S.; Kendall, D.; Krauss, R.M.; et al. American College of Endocrinology position statement on the insulin resistance syndrome. Endocr. Pract. 2003, 9, 237–252. [Google Scholar] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Ferri, N.; Ruscica, M. Proprotein convertase subtilisin/kexin type 9 (PCSK9) and metabolic syndrome: Insights on insulin resistance, inflammation, and atherogenic dyslipidemia. Endocrine 2016, 54, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z.; Motro, M.; Adler, Y. Atherogenic dyslipidemia in metabolic syndrome and type 2 diabetes: Therapeutic options beyond statins. Cardiovasc. Diabetol. 2006, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Redfern, J.S.; McGovern, M.E.; Giral, P. Niacin and fibrates in atherogenic dyslipidemia: Pharmacotherapy to reduce cardiovascular risk. Pharmacol. Ther. 2010, 126, 314–345. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog. Mol. Biol. Transl. Sci. 2014, 121, 217–266. [Google Scholar] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the safety of PPAR agonists—Current trends and future prospects. Expert Opin. Drug Saf. 2013, 12, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Bellosta, S.; Davidson, M.H. Pharmacokinetic interactions between statins and fibrates. Am. J. Cardiol. 2005, 96, 44K–49K; discussion 34K–35K. [Google Scholar] [CrossRef] [PubMed]
- Magni, P.; Macchi, C.; Morlotti, B.; Sirtori, C.R.; Ruscica, M. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur. J. Intern. Med. 2015, 26, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, G. PPARs Integrate the Mammalian Clock and Energy Metabolism. PPAR Res. 2014, 2014, 653017. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1996, 1302, 93–109. [Google Scholar] [CrossRef]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Saluja, I.; Granneman, J.G.; Skoff, R.P. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 2001, 33, 191–204. [Google Scholar] [CrossRef]
- Cermenati, G.; Audano, M.; Giatti, S.; Carozzi, V.; Porretta-Serapiglia, C.; Pettinato, E.; Ferri, C.; D’Antonio, M.; De Fabiani, E.; Crestani, M.; et al. Lack of sterol regulatory element binding factor-1c imposes glial Fatty Acid utilization leading to peripheral neuropathy. Cell Metab. 2015, 21, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [PubMed]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kim, M.; Park, H.S.; Kim, H.S.; Jeon, M.J.; Oh, K.S.; Koh, E.H.; Won, J.C.; Kim, M.S.; Oh, G.T.; et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem. Biophys. Res. Commun. 2006, 340, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Peters, J.M.; Harris, R.A. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem. Biophys. Res. Commun. 2001, 287, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Wahli, W. Peroxisome proliferator-activated receptors A link between endocrinology and nutrition? Trends Endocrinol. Metab. 1993, 4, 291–296. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Gervois, P.P.; Verschuren, L.; Staels, B.; Princen, H.M.; Kooistra, T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NFkappa B-C/EBP-beta complex formation. Blood 2003, 101, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.R.; Clarke, S.; Rodgers, K.; Thornhill, B.; Peters, J.M.; Gonzalez, F.J.; Gimble, J.M. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect. Immun. 1999, 67, 3488–3493. [Google Scholar] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Amri, E.Z.; Bonino, F.; Ailhaud, G.; Abumrad, N.A.; Grimaldi, P.A. Cloning of a protein that mediates transcriptional effects of fatty acids in preadipocytes. Homology to peroxisome proliferator-activated receptors. J. Biol. Chem. 1995, 270, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Hertz, R.; Berman, I.; Keppler, D.; Bar-Tana, J. Activation of gene transcription by prostacyclin analogues is mediated by the peroxisome-proliferators-activated receptor (PPAR). Eur. J. Biochem. 1996, 235, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor delta in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, K.S.; Billin, A.N. PPARbeta/delta ligands as modulators of the inflammatory response. Curr. Opin. Investig. Drugs 2008, 9, 463–469. [Google Scholar] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, Y.; Tang, Z.; Zhang, H.; Qin, X.; Zhu, Y.; Guan, Y.; Wang, X.; Staels, B.; Chien, S.; et al. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Piraino, G.; Hake, P.W.; O’Connor, M.; Denenberg, A.; Fan, H.; Cook, J.A. Peroxisome proliferator-activated receptor {delta} regulates inflammation via NF-{kappa}B signaling in polymicrobial sepsis. Am. J. Pathol. 2010, 177, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARgamma in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Zhang, X.; Zhu, C.; Tang, X.; Yu, F.; Shang, G.W.; Cai, X. Molecular Mechanisms of PPAR-gamma Governing MSC Osteogenic and Adipogenic Differentiation. Curr. Stem Cell Res. Ther. 2016, 11, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yoshioka, S.; Yoshioka, T.; Ushiyama, I.; Horikoshi, H. Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes 1988, 37, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J. Clin. Investig. 2000, 106, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Miles, P.D.; Barak, Y.; He, W.; Evans, R.M.; Olefsky, J.M. Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J. Clin. Investig. 2000, 105, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Steger, D.J.; Zhuo, D.; Qatanani, M.; Mullican, S.E.; Tuteja, G.; Manduchi, E.; Grant, G.R.; Lazar, M.A. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol. Cell. Biol. 2010, 30, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Foryst-Ludwig, A.; Hartge, M.; Clemenz, M.; Sprang, C.; Hess, K.; Marx, N.; Unger, T.; Kintscher, U. PPARgamma activation attenuates T-lymphocyte-dependent inflammation of adipose tissue and development of insulin resistance in obese mice. Cardiovasc. Diabetol. 2010, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Motoki, T.; Kurobe, H.; Hirata, Y.; Nakayama, T.; Kinoshita, H.; Rocco, K.A.; Sogabe, H.; Hori, T.; Sata, M.; Kitagawa, T. PPAR-gamma agonist attenuates inflammation in aortic aneurysm patients. Gen. Thorac. Cardiovasc. Surg. 2015, 63, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Ginty, M.; Kemp, K.; Scolding, N.; Wilkins, A. The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J. Neuroinflammation 2012, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.; Castellino, G.; Banach, M.; Toth, P.P.; Ivanova, E.; Orekhov, A.N.; Montalto, G.; Rizzo, M. PPAR Agonists, Atherogenic Dyslipidemia and Cardiovascular Risk. Curr. Pharm. Des. 2017, 23, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Dash, S.; Morgantini, C.; Hegele, R.A.; Lewis, G.F. Pharmacological Targeting of the Atherogenic Dyslipidemia Complex: The Next Frontier in CVD Prevention Beyond Lowering LDL Cholesterol. Diabetes 2016, 65, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.A.; Kizhakepunnur, L.G.; Bahekar, A.; Arora, R.R. The role of fibrates in the prevention of cardiovascular disease—A pooled meta-analysis of long-term randomized placebo-controlled clinical trials. Am. Heart J. 2007, 154, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Sirtori, C.R. Mechanisms of action of absorbable hypolipidemic drugs. Adv. Exp. Med. Biol. 1985, 183, 241–252. [Google Scholar] [PubMed]
- Sirtori, C.R. The FIELD study. Lancet 2006, 367, 1141–1142; author reply 1142–1143. [Google Scholar] [CrossRef]
- Elam, M.B.; Ginsberg, H.N.; Lovato, L.C.; Corson, M.; Largay, J.; Leiter, L.A.; Lopez, C.; O’Connor, P.J.; Sweeney, M.E.; Weiss, D.; et al. Association of Fenofibrate Therapy With Long-term Cardiovascular Risk in Statin-Treated Patients With Type 2 Diabetes. JAMA Cardiol. 2017, 2, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A.; Sirtori, C.; Ruscica, M. PPAR-alpha agonists are still on the rise: An update on clinical and experimental findings. Expert Opin. Investig. Drugs 2017, 26, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Simental-Mendia, L.E.; Simental-Mendia, M.; Sanchez-Garcia, A.; Banach, M.; Atkin, S.L.; Gotto, A.M., Jr.; Sahebkar, A. Effect of fibrates on glycemic parameters: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol. Res. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Latruffe, N. Medical significance of peroxisome proliferator-activated receptors. Lancet 1999, 354, 141–148. [Google Scholar] [CrossRef]
- Brown, J.D.; Plutzky, J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Flory, J.H.; Ellenberg, S.; Szapary, P.O.; Strom, B.L.; Hennessy, S. Antidiabetic action of bezafibrate in a large observational database. Diabetes Care 2009, 32, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome Analysis of K-877 (a Novel Selective PPARalpha Modulator (SPPARMalpha))-Regulated Genes in Primary Human Hepatocytes and the Mouse Liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a Novel Selective PPARalpha Modulator, on Lipid and Glucose Metabolism in Patients With Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendia, L.E.; Mikhailidis, D.P.; Pirro, M.; Banach, M.; Sirtori, C.R.; Ruscica, M.; Reiner, Z. Effect of statin therapy on plasma apolipoprotein CIII concentrations: A systematic review and meta-analysis of randomized controlled trials. J. Clin. Lipidol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S.; Group, K.S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor alpha modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G. Phase 2 clinical trials with K-877 (pemafibrate): A promising selective PPAR-alpha modulator for treatment of combined dyslipidemia. Atherosclerosis 2017, 261, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [PubMed]
- Fruchart, J.C. Pemafibrate (K-877), a novel selective peroxisome proliferator-activated receptor alpha modulator for management of atherogenic dyslipidaemia. Cardiovasc. Diabetol. 2017, 16, 124. [Google Scholar] [CrossRef] [PubMed]
- NCT03071692. Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts with diabeTes (PROMINENT). Available online: https://www.clinicaltrials.gov/ct2/results?cond=&term=NCT03071692&cntry=&state=&city=&dist= (accessed on 13 April 2018).
- Sirtori, C.R.; Galli, C.; Franceschini, G. Fraudulent (and non fraudulent) fatty acids for human health. Eur. J. Clin. Investig. 1993, 23, 686–689. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jia, Y.; Fu, T.; Viswakarma, N.; Bai, L.; Rao, M.S.; Zhu, Y.; Borensztajn, J.; Reddy, J.K. Sustained activation of PPARalpha by endogenous ligands increases hepatic fatty acid oxidation and prevents obesity in ob/ob mice. FASEB J. 2012, 26, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Update on the management of severe hypertriglyceridemia—Focus on free fatty acid forms of omega-3. Drug Des. Dev. Ther. 2015, 9, 2129–2137. [Google Scholar]
- Pirillo, A.; Catapano, A.L. Omega-3 polyunsaturated fatty acids in the treatment of hypertriglyceridaemia. Int. J. Cardiol. 2013, 170 (Suppl. S1), S16–S20. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.F.; Burke, F.M.; Soffer, D.E. Review of Cardiometabolic Effects of Prescription Omega-3 Fatty Acids. Curr. Atheroscler. Rep. 2017, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Sperling, L.S.; Nelson, J.R. History and future of omega-3 fatty acids in cardiovascular disease. Curr. Med. Res. Opin. 2016, 32, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Pizzini, A.; Lunger, L.; Demetz, E.; Hilbe, R.; Weiss, G.; Ebenbichler, C.; Tancevski, I. The Role of Omega-3 Fatty Acids in Reverse Cholesterol Transport: A Review. Nutrients 2017, 9, 1099. [Google Scholar] [CrossRef] [PubMed]
- Pahlavani, M.; Ramalho, T.; Koboziev, I.; LeMieux, M.J.; Jayarathne, S.; Ramalingam, L.; Filgueiras, L.R.; Moustaid-Moussa, N. Adipose tissue inflammation in insulin resistance: Review of mechanisms mediating anti-inflammatory effects of omega-3 polyunsaturated fatty acids. J. Investig. Med. 2017, 65, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Rossmeisl, M.; Kopecky, J. Omega-3 fatty acids and adipose tissue biology. Mol. Aspects Med. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.U.; Ohmori, K.; Hashimoto, T.; Kamitori, K.; Yamaguchi, F.; Noma, T.; Igarashi, J.; Tsuboi, K.; Tokuda, M.; Nishiyama, A.; et al. GPR120 in adipocytes has differential roles in the production of pro-inflammatory adipocytokines. Biochem. Biophys. Res. Commun. 2017, 486, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Bargut, T.C.; Silva-e-Silva, A.C.; Souza-Mello, V.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Mice fed fish oil diet and upregulation of brown adipose tissue thermogenic markers. Eur. J. Nutr. 2016, 55, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Lopez, T.; Cereijo, R.; Turatsinze, J.V.; Planavila, A.; Cairo, M.; Gavalda-Navarro, A.; Peyrou, M.; Moure, R.; Iglesias, R.; Giralt, M.; et al. The lipid sensor GPR120 promotes brown fat activation and FGF21 release from adipocytes. Nat. Commun. 2016, 7, 13479. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, A.J.; Cruz, A.L.; Heyward, W.L.; Bulkow, L.R.; Hall, D.; Barstaed, L.; Connor, W.E. Elevated concentrations of plasma omega-3 polyunsaturated fatty acids among Alaskan Eskimos. Am. J. Clin. Nutr. 1994, 59, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Park, H.G.; Zhang, J.Y.; Lawrence, P.; Liu, G.; Subramanian, N.; Kothapalli, K.S.; Brenna, J.T. Brown but not white adipose cells synthesize omega-3 docosahexaenoic acid in culture. Prostaglandins Leukot. Essent. Fatty Acids 2016, 104, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Baragetti, A.; Catapano, A.L.; Norata, G.D. Translating the biology of adipokines in atherosclerosis and cardiovascular diseases: Gaps and open questions. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Liddle, D.M.; De Boer, A.A.; Brown, M.J.; Power, K.A.; Ma, D.W.; Robinson, L.E. Fish-oil-derived n-3 PUFAs reduce inflammatory and chemotactic adipokine-mediated cross-talk between co-cultured murine splenic CD8+ T cells and adipocytes. J. Nutr. 2015, 145, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Ballantyne, C.M.; Braeckman, R.A.; Stirtan, W.G.; Doyle, R.T., Jr.; Philip, S.; Soni, P.N.; Juliano, R.A. Icosapent Ethyl (Eicosapentaenoic Acid Ethyl Ester): Effects Upon High-Sensitivity C-Reactive Protein and Lipid Parameters in Patients With Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Buoite Stella, A.; Gortan Cappellari, G.; Barazzoni, R.; Zanetti, M. Update on the Impact of Omega 3 Fatty Acids on Inflammation, Insulin Resistance and Sarcopenia: A Review. Int. J. Mol. Sci. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.Y.; Jacobson, T.A. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: A systematic review and meta-analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, J. Omega-3 Fatty Acid Formulations in Cardiovascular Disease: Dietary Supplements are Not Substitutes for Prescription Products. Am. J. Cardiovasc. Drugs 2016, 16, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.G.; Susekov, A.V.; de Bruin, T.W.A.; Kvarnstrom, M.; Yang, H.; Davidson, M.H. Omega-3 carboxylic acids in patients with severe hypertriglyceridemia: EVOLVE II, a randomized, placebo-controlled trial. J. Clin. Lipidol. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.; Maki, K.C.; Susekov, A.; Ezhov, M.; Nordestgaard, B.G.; Machielse, B.N.; Kling, D.; Davidson, M.H. Omega-3 free fatty acids for the treatment of severe hypertriglyceridemia: The EpanoVa fOr Lowering Very high triglyceridEs (EVOLVE) trial. J. Clin. Lipidol. 2014, 8, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Orloff, D.G.; Nicholls, S.J.; Dunbar, R.L.; Roth, E.M.; Curcio, D.; Johnson, J.; Kling, D.; Davidson, M.H. A highly bioavailable omega-3 free fatty acid formulation improves the cardiovascular risk profile in high-risk, statin-treated patients with residual hypertriglyceridemia (the ESPRIT trial). Clin. Ther. 2013, 35, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 1999, 354, 447–455. [Google Scholar]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Kromhout, D.; Giltay, E.J.; Geleijnse, J.M.; Alpha Omega Trial Group. n-3 fatty acids and cardiovascular events after myocardial infarction. N. Engl. J. Med. 2010, 363, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Halsey, J.; Kromhout, D.; Gerstein, H.C.; Marchioli, R.; Tavazzi, L.; Geleijnse, J.M.; Rauch, B.; Ness, A.; Galan, P.; et al. Associations of Omega-3 Fatty Acid Supplement Use With Cardiovascular Disease Risks: Meta-analysis of 10 Trials Involving 77917 Individuals. JAMA Cardiol. 2018, 3, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Brinton, E.A.; Jacobson, T.A.; Miller, M.; Tardif, J.C.; Ketchum, S.B.; Doyle, R.T., Jr.; Murphy, S.A.; Soni, P.N.; et al. Rationale and design of REDUCE-IT: Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial. Clin. Cardiol. 2017, 40, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Baldessin, L.; Boccia, D.; Racagni, G.; Mitro, N. Non-insulin anti-diabetic drugs: An update on pharmacological interactions. Pharmacol. Res. 2017, 115, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Rosiglitazone revisited: An updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med. 2010, 170, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Wolski, K.; Nicholls, S.J.; Nissen, S.E. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: A meta-analysis of randomized trials. JAMA 2007, 298, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Betteridge, D.J. CHICAGO, PERISCOPE and PROactive: CV risk modification in diabetes with pioglitazone. Fundam. Clin. Pharmacol. 2009, 23, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Nicholls, S.J.; Wolski, K.; Nesto, R.; Kupfer, S.; Perez, A.; Jure, H.; De Larochelliere, R.; Staniloae, C.S.; Mavromatis, K.; et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 2008, 299, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Tuzcu, E.M.; Wolski, K.; Bayturan, O.; Lavoie, A.; Uno, K.; Kupfer, S.; Perez, A.; Nesto, R.; Nissen, S.E. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: Insights from the PERISCOPE (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation) study. J. Am. Coll. Cardiol. 2011, 57, 153–159. [Google Scholar] [PubMed]
- Mazzone, T.; Meyer, P.M.; Feinstein, S.B.; Davidson, M.H.; Kondos, G.T.; D’Agostino, R.B., Sr.; Perez, A.; Provost, J.C.; Haffner, S.M. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 2006, 296, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.; Meyer, P.M.; Haffner, S.; Feinstein, S.; D’Agostino, R., Sr.; Kondos, G.T.; Perez, A.; Chen, Z.; Mazzone, T. Increased high-density lipoprotein cholesterol predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with type 2 diabetes mellitus. Circulation 2008, 117, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Viscoli, C.M.; Young, L.H.; Furie, K.L.; Gorman, M.; Lovejoy, A.M.; Dagogo-Jack, S.; Ismail-Beigi, F.; Korytkowski, M.T.; Pratley, R.E.; et al. Pioglitazone Prevents Diabetes in Patients With Insulin Resistance and Cerebrovascular Disease. Diabetes Care 2016, 39, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Viscoli, C.M.; Inzucchi, S.E.; Young, L.H.; Insogna, K.L.; Conwit, R.; Furie, K.L.; Gorman, M.; Kelly, M.A.; Lovejoy, A.M.; Kernan, W.N.; et al. Pioglitazone and Risk for Bone Fracture: Safety Data from a Randomized Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARgamma signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Pena, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vazquez-Carrera, M. PPARbeta/delta: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Majumder, A.; Ray, S. Observational study of effects of Saroglitazar on glycaemic and lipid parameters on Indian patients with type 2 diabetes. Sci. Rep. 2015, 5, 7706. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [PubMed]
- Balakumar, P.; Rose, M.; Ganti, S.S.; Krishan, P.; Singh, M. PPAR dual agonists: Are they opening Pandora’s Box? Pharmacol. Res. 2007, 56, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Pavanello, C.; Calabresi, L.; Ruscica, M. Nutraceutical approaches to metabolic syndrome. Ann. Med. 2017, 49, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Unmet need for primary prevention in individuals with hypertriglyceridaemia not eligible for statin therapy according to European Society of Cardiology/European Atherosclerosis Society guidelines: A contemporary population-based study. Eur. Heart J. 2018, 39, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol. Res. 2018, 128, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Nordestgaard, B.G. Is It Time for New Thinking about High-Density Lipoprotein? Arterioscler. Thromb. Vasc. Biol. 2018, 38, 484–486. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Major roles of different peroxisome proliferator-activated receptors (PPARs) isotypes. PPARs are a class of nuclear transcription factors that heterodimerize with retinoid X receptor (RXR, gray boxes) upon physiological (i.e., fatty acids) and synthetic activation (i.e., fibrates, glitazones etc.) to regulate the specific indicated pathways. FA, Fatty Acids; NFκB, Nuclear Factor-κB.
Figure 1.
Major roles of different peroxisome proliferator-activated receptors (PPARs) isotypes. PPARs are a class of nuclear transcription factors that heterodimerize with retinoid X receptor (RXR, gray boxes) upon physiological (i.e., fatty acids) and synthetic activation (i.e., fibrates, glitazones etc.) to regulate the specific indicated pathways. FA, Fatty Acids; NFκB, Nuclear Factor-κB.


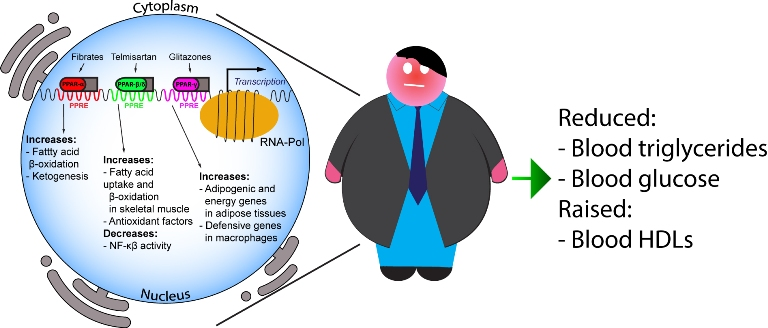{kind=link}
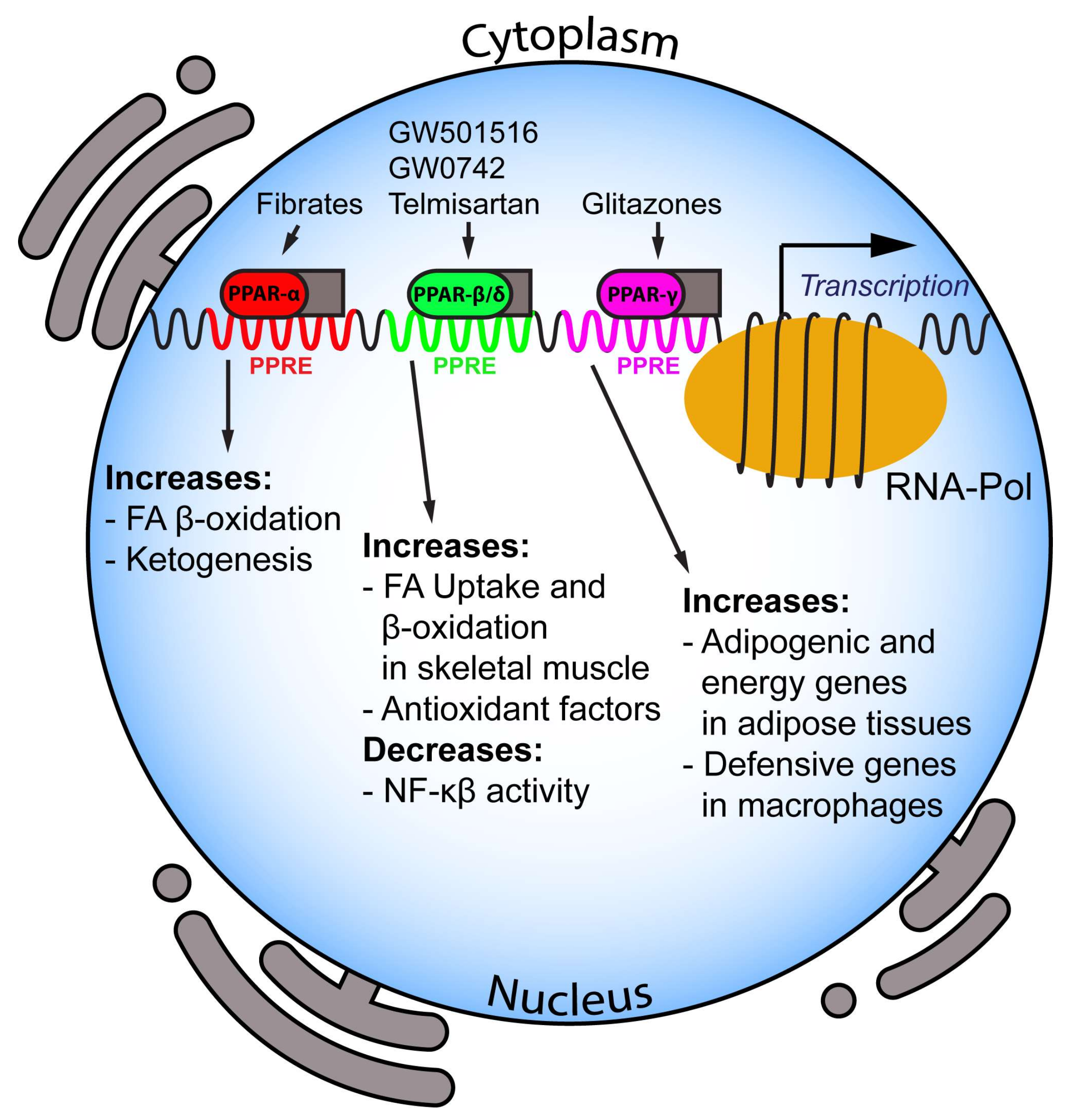{kind=link}
Table 1.
Risk factors for the clinical diagnosis of metabolic syndrome.
| Value | Alternative Indicator | |
|---|---|---|
| Waist circumference | * >94 cm in males, >80 cm in females ** >102 cm in males, >88 cm in females | |
| Raised blood pressure | Systolic ≥130 and/or diastolic ≥85 mm Hg | Treatment of previously diagnosed hypertension |
| Raised FPG | ≥100 mg/dL (5.6 mmol/L) | Previously diagnosed of T2DM |
| Raised TG | >150 mg/dL (1.7 mmol/L) | Specific pharmacological treatment |
| Reduced HDL-C | <40 mg/dL (1.0 mmol/L) in males <50 mg/dL (1.3 mmol/L) in females | Specific pharmacological treatment |
* Based on the International Diabetes Federation (IDF) threshold for Europid population. ** Based on the AHA/NHLBI (ATP III) threshold for USA population. FPG, Fasting Plasma Glucose; TG, triglycerides; HDL-C, High-Density Lipoprotein-Cholesterol; T2DM, Type 2 Diabetes Mellitus. Conversion factors: (i) mg/dL cholesterol = mmol/L × 38.6; (ii) mg/dL triglycerides = mmol/L × 88.5 and (iii) mg/dL glucose = mmol/L × 18. Reproduced with permission [8].
Table 2.
Effect of PPARs on the features of metabolic syndrome—evidence from clinical trials.
| PPAR-α Agonist | Clinic Study | Major Findings |
|---|---|---|
| Pemafibrate | Phase 3 (JapicCTI-142412; clinicaltrials.jp) follow-up: 24 weeks subjects: 166 [74] | 1. Reduction in TGs: −45% 2. Decrement in non-HDL 3. Increase in HDL cholesterol |
| Phase 3 (JapicCTI-142620; clinicaltrials.jp) follow-up: 24 weeks subjects: 225 [76] | 1. Reduction in TGs: −46.2% 2. A further −6.5% TG reduction compared to fenofibrate | |
| Phase 3 (JapicCTI-121764; clinicaltrials.jp) follow-up: 12 weeks subjects: 489 [77] | 1. TGs: −46.3% (0.1 mg/day), −46.7% (0.2 mg/day) and −51.8% (0.4 mg/day) vs. −38.3% (fenofibrate 100 mg/day) and −51.5% (fenofibrate 200 mg/day) | |
| Phase 2 follow-up: 12 weeks subjects: 188 [79] | 1. Reduction in TGs: range from −46.1% to −53.4% | |
| Phase 2 follow-up: 24 weeks subjects: 423 [79] | 1. Reduction in TGs: range from −46.8% to −50.8% | |
| On going Phase 3 trial PROMINENT (Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts with diabeTes)—NCT03071692 | Outcomes: First occurrence of nonfatal myocardial infarction, nonfatal ischemic stroke, hospitalization for unstable angina requiring unplanned coronary revascularization, and CV death. | |
| Omega-3 | EVOLVEII (Epanova® for Lowering Very High Triglycerides II)—NCT02009865 Phase 3 follow-up: 12 weeks subjects: 162 [106] | 1. Reduction in TGs: −14.2% 2. Reduction in non-HDL-C: −9% |
| EVOLVE (The EpanoVa fOr Lowering Very high triglyceridEs)—NCT01242527 Phase 3 follow-up: 12 weeks subjects: 399 [107] | 1. Reduction in TGs: range −25.5%/−30.9% 2. Reduction in non-HDL-C: range from −6.9% to −9.6% | |
| ESPRIT (EPANOVA Combined with a STATIN in PATIENTS With HYPERTRIGLYCERIDEMIA to Reduce Non-HDL CHOLESTEROL)—NCT01408303. Phase 3 follow-up: 6 weeks subjects: 647 [108] | 1. Reduction in TGs: range from −14.6% to −20.6% 2. Reduction in non-HDL-C: range from −3.9% to −6.9% | |
| On going phase 3 trials: (i) STRENGTH (Study to assess statin residual risk Reduction with Epanova in high cardiovascular risk patients with Hypertriglyceridemia)—NCT02104817 (ii) REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention)—NCT01492361 [113] | Outcomes: First occurrence of cardiovascular death, nonfatal MI, nonfatal stroke, emergent/elective coronary revascularization, or hospitalization for unstable angina | |
| Pioglitazone | The PERISCOPE Trial (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation)—NCT00225277 Phase 3 follow-up: 18 months subjects: 543 [119] | 1. Percent atheroma volume change: −0.16% 2. Raise in HDL-C: +5.7 mg/dL 3. Decrement in TGs: −16.3 mg/dL |
| The CHICAGO (Carotid Intima-Media Thickness in Atherosclerosis Using Pioglitazone) trial—NCT00225264 Phase 3 follow-up: 72 weeks subjects: 462 [121,122] | 1. Progression of mean CIMT: −0.013 mm vs. glimepiride 2. Progression of maximum CIMT: −0.024 mm vs. glimepiride 3. HDL-C: +14% | |
| The IRIS (Insulin Resistance Intervention after Stroke)—NCT00091949 Phase 3 follow-up: 4.8 years subjects: 3876 [123,124] | 1. Reduction of stroke or MI in insulin resistant patients 2. Reduction in recurrence of diabetes: −52% | |
| Elafibranor | GOLDEN trial—NCT01694849 Phase 2b follow-up: 52 weeks subjects: 256 [131] | 1. NASH resolution in 19% of patients |
All percentage changes are vs. baseline otherwise differently indicated. CIMT, Carotid Intima-Media Thickness; CV, cardiovascular; HDL-C, high-density lipoprotein cholesterol; MI, Miocardial Infarction; NASH, nonalcoholic steatohepatitis; TG, triglyceride.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. https://doi.org/10.3390/ijms19041197
AMA Style
Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M. PPAR Agonists and Metabolic Syndrome: An Established Role? International Journal of Molecular Sciences. 2018; 19(4):1197. https://doi.org/10.3390/ijms19041197
Chicago/Turabian StyleBotta, Margherita, Matteo Audano, Amirhossein Sahebkar, Cesare R. Sirtori, Nico Mitro, and Massimiliano Ruscica. 2018. "PPAR Agonists and Metabolic Syndrome: An Established Role?" International Journal of Molecular Sciences 19, no. 4: 1197. https://doi.org/10.3390/ijms19041197
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.