Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication

 ,
,  ,
,
Abstract
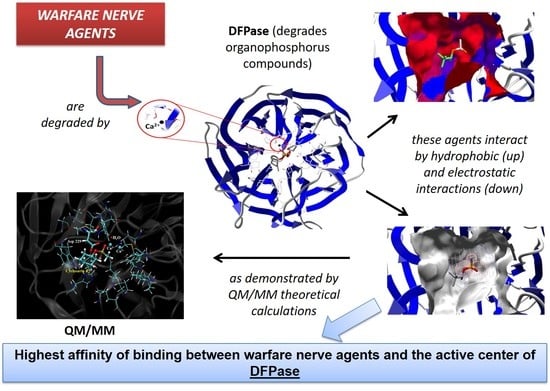
:
1. Introduction
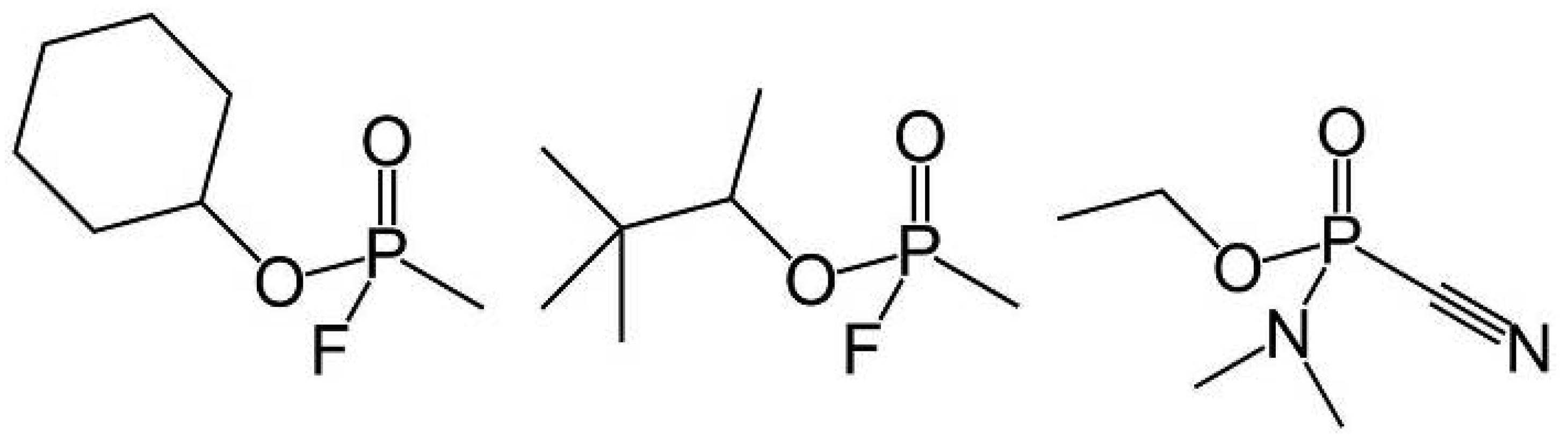
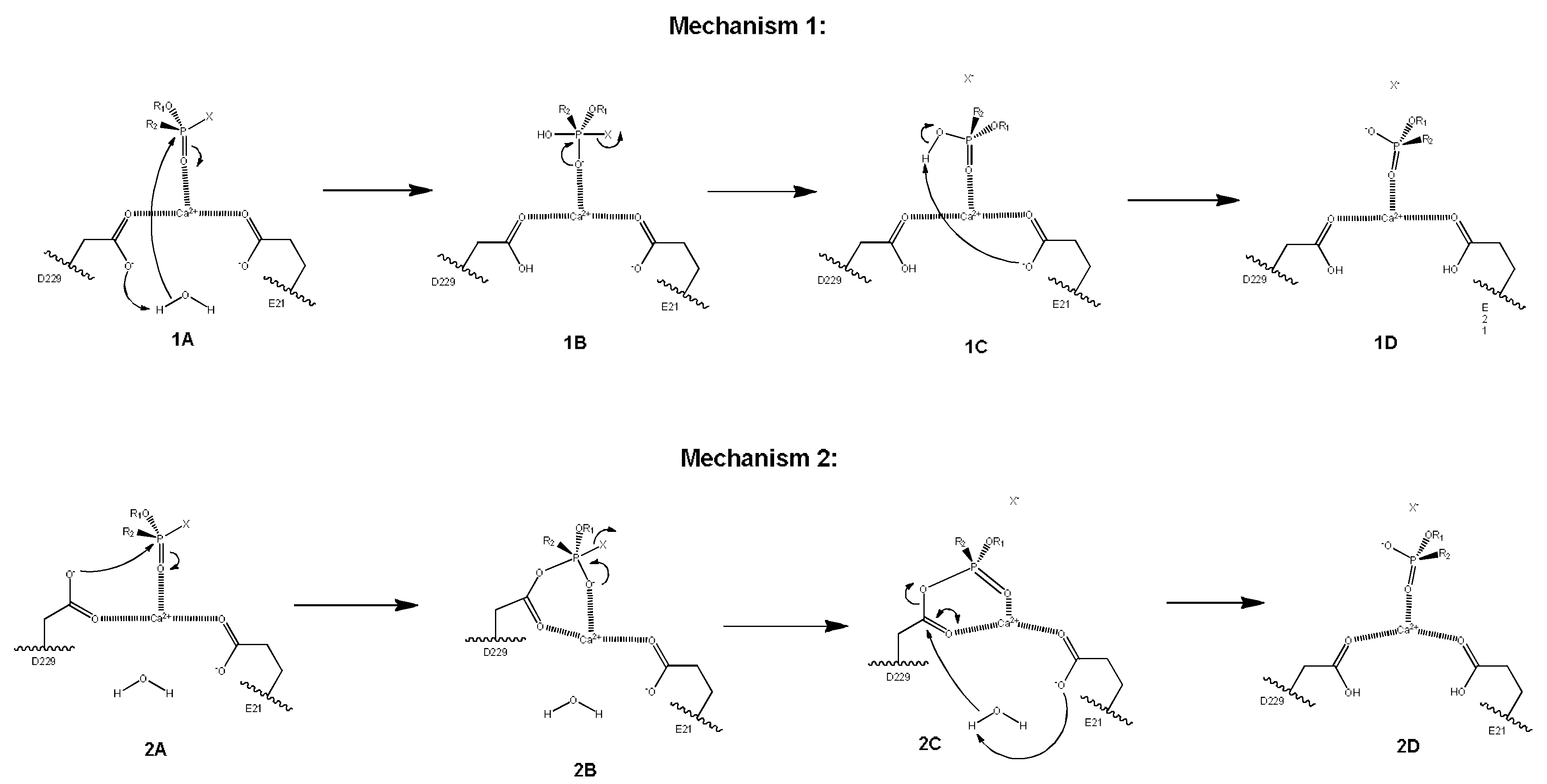
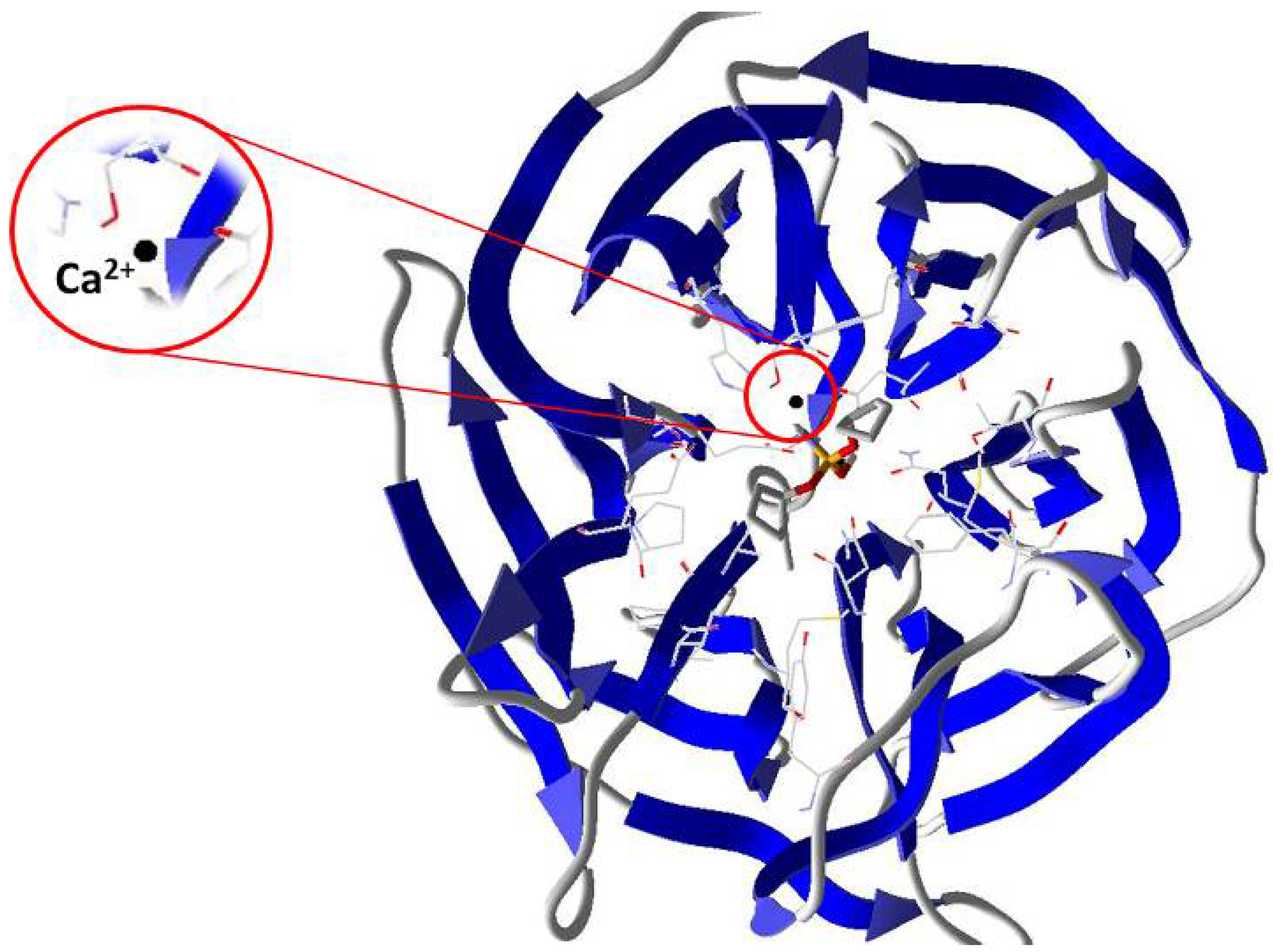
2. Results and Discussion
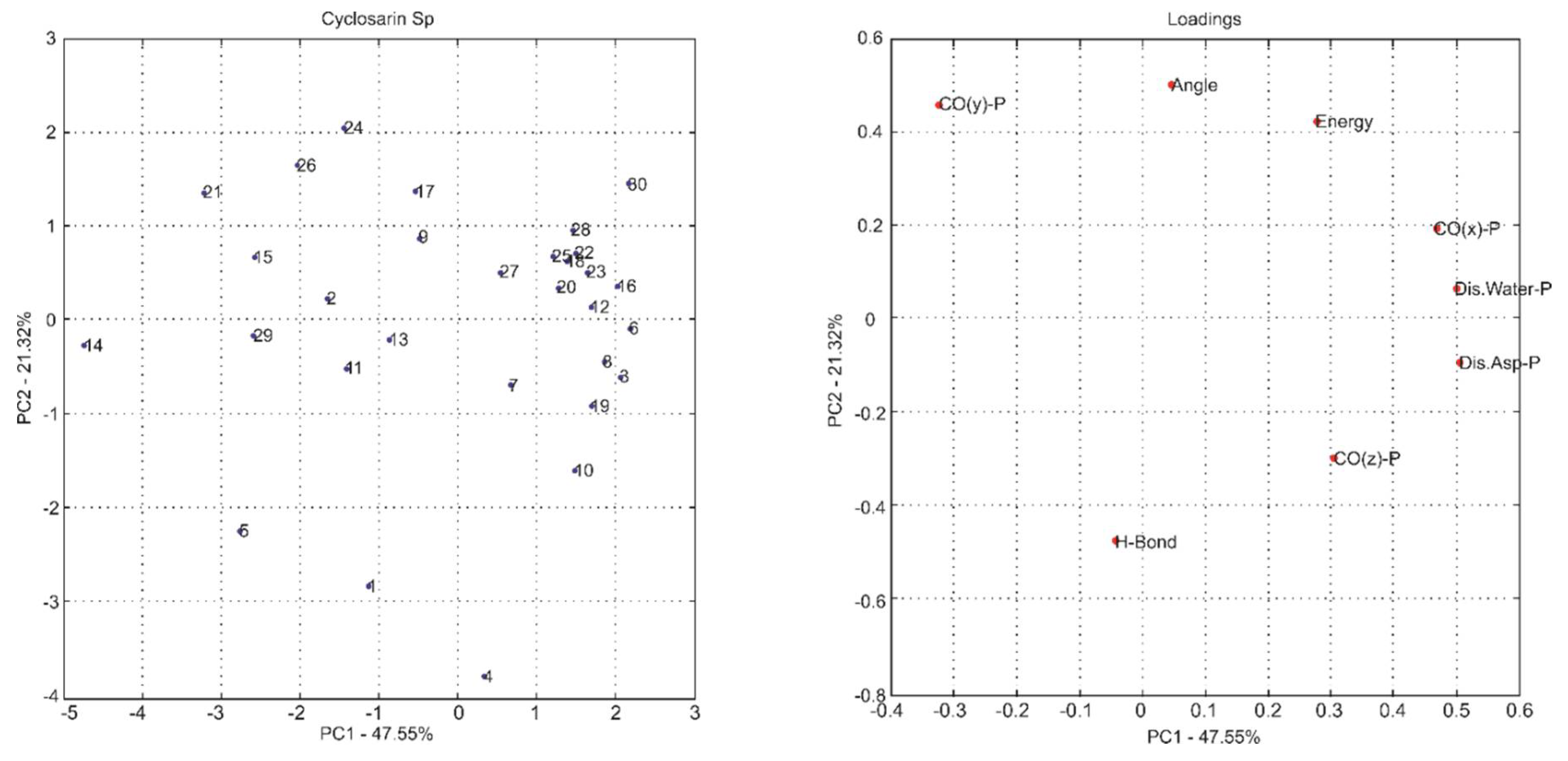
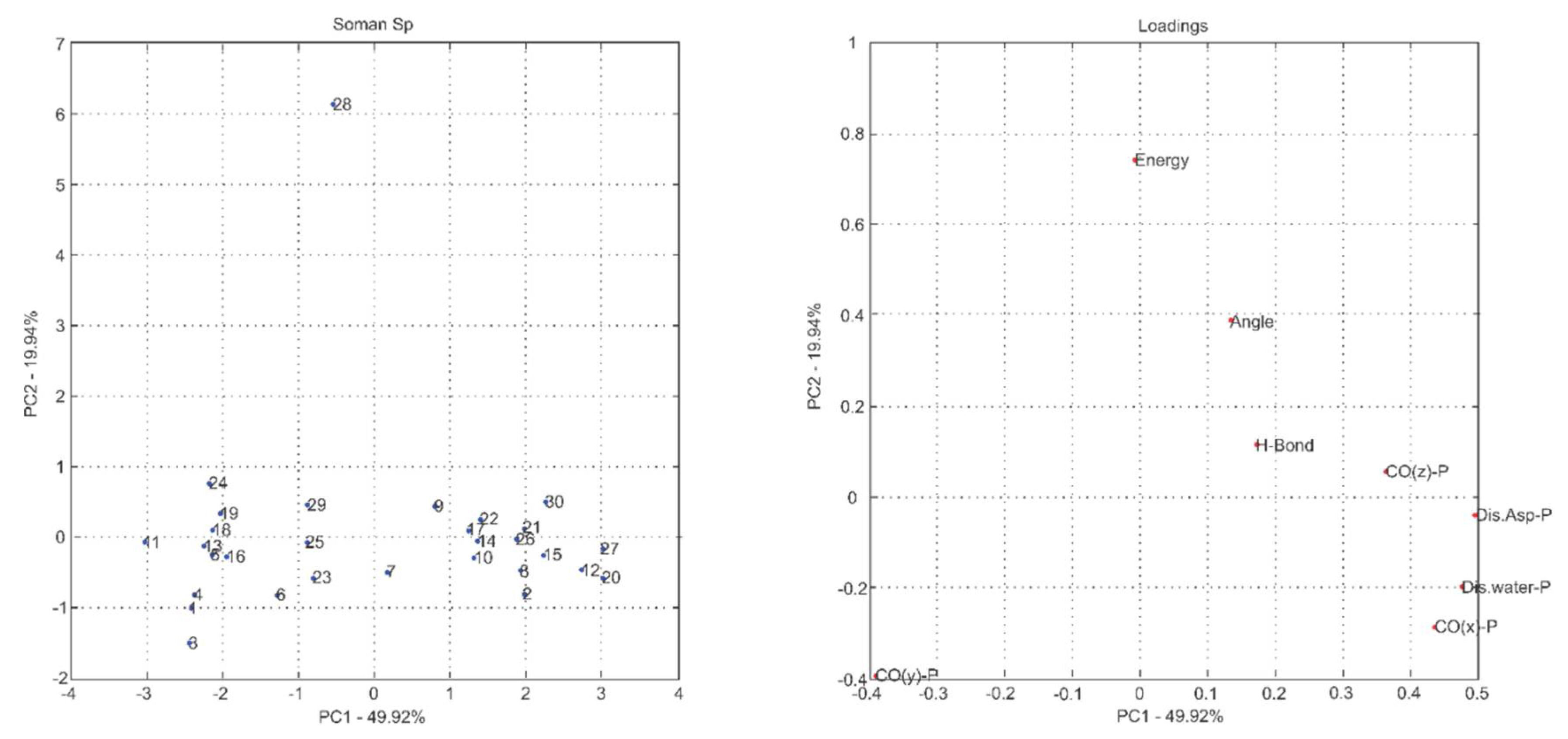
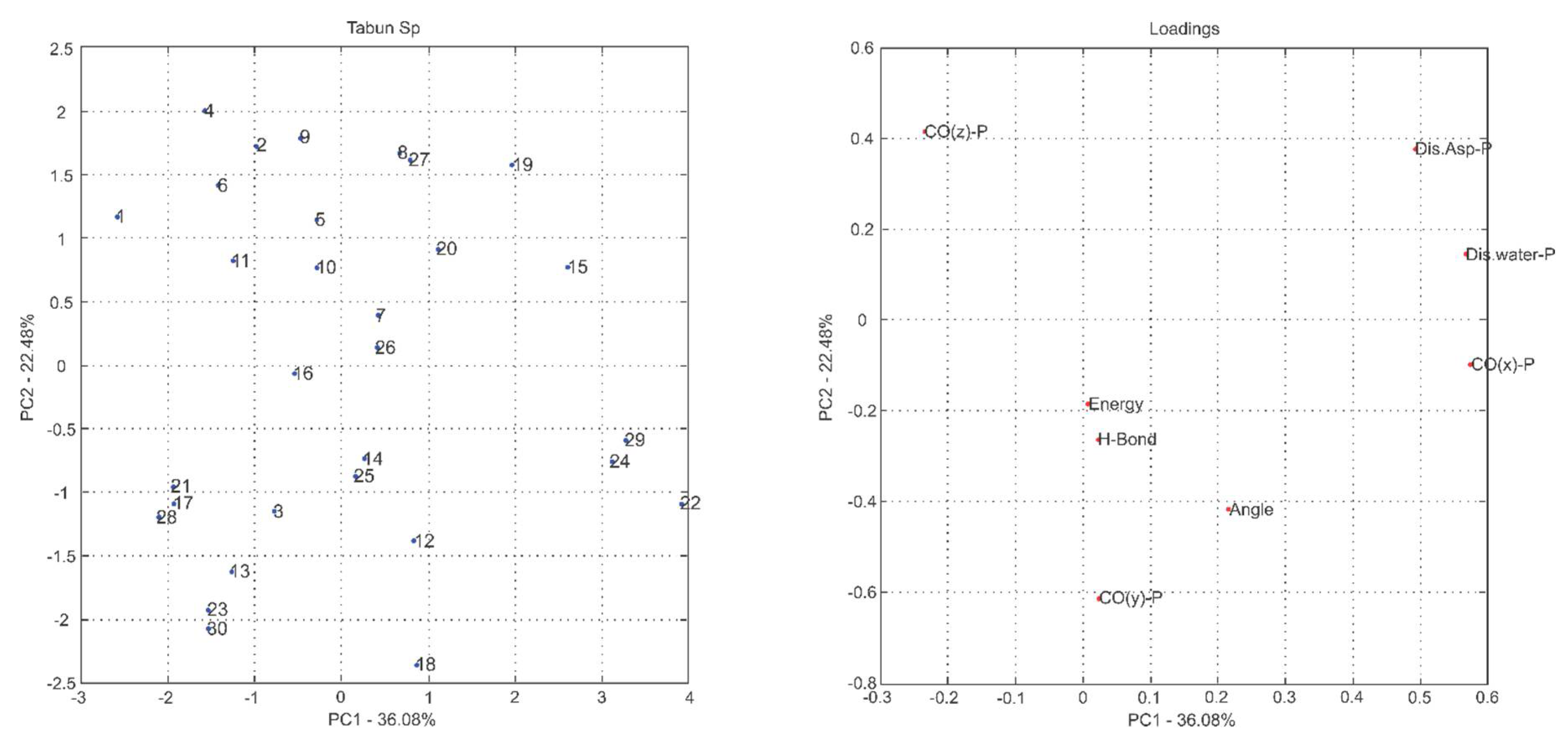
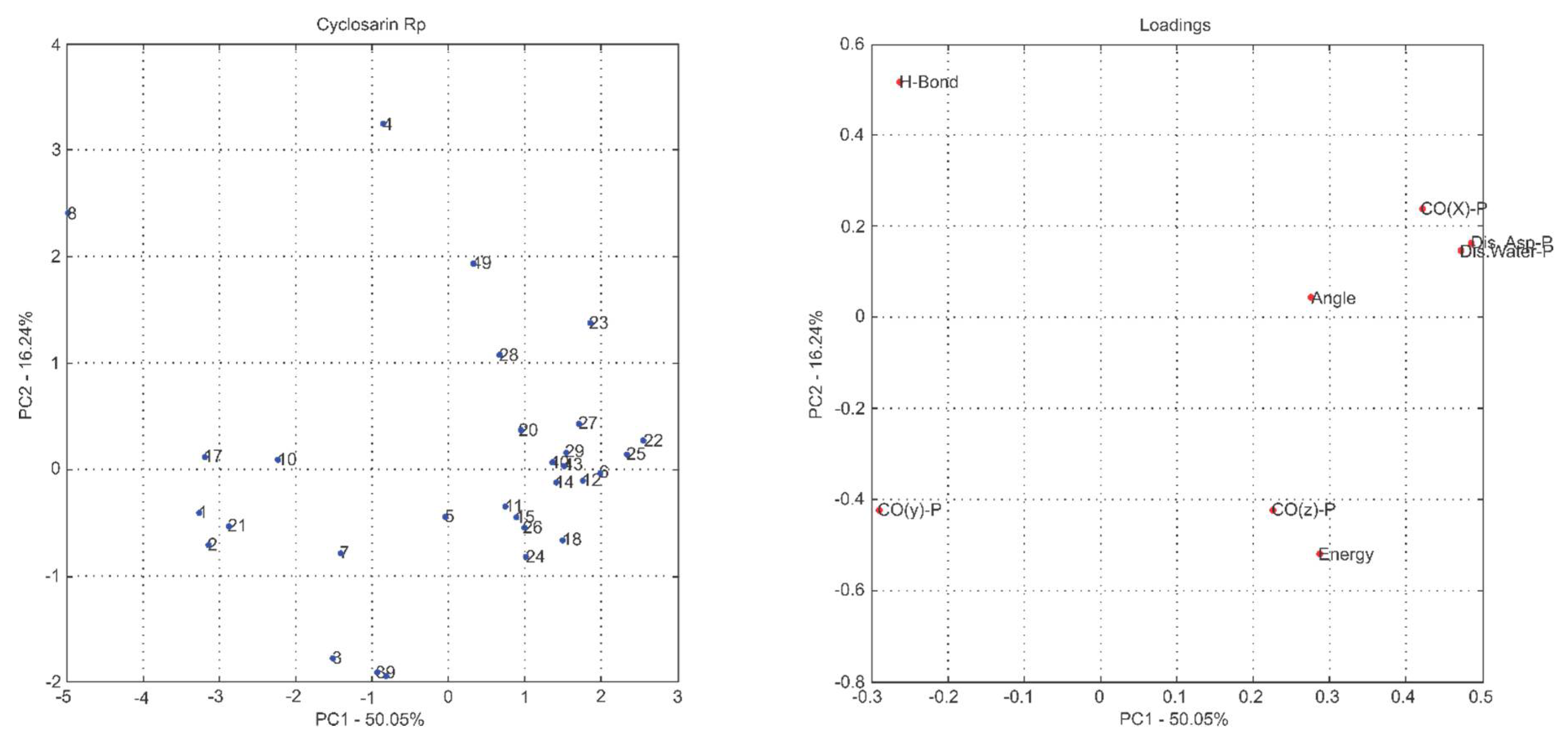
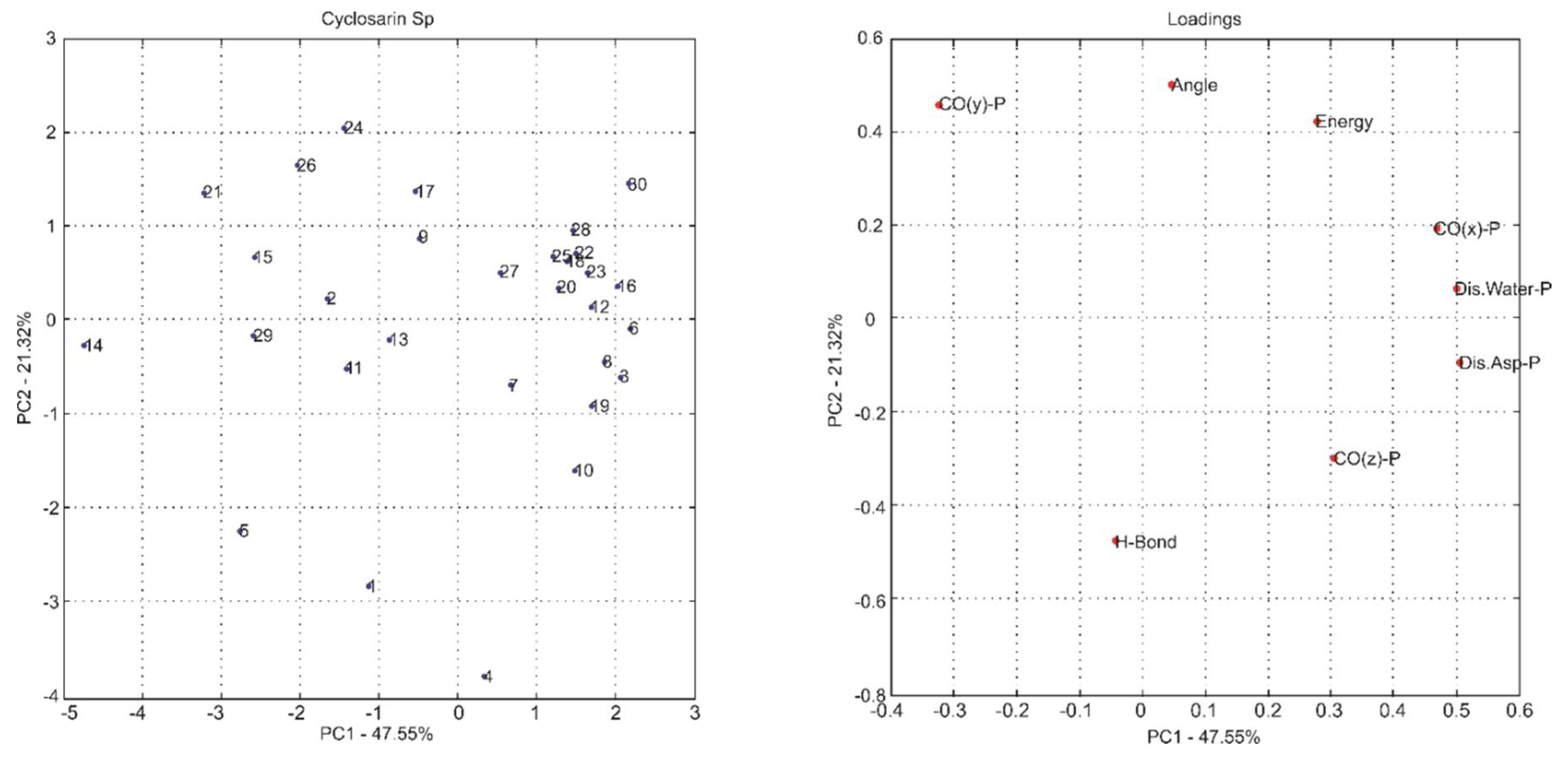
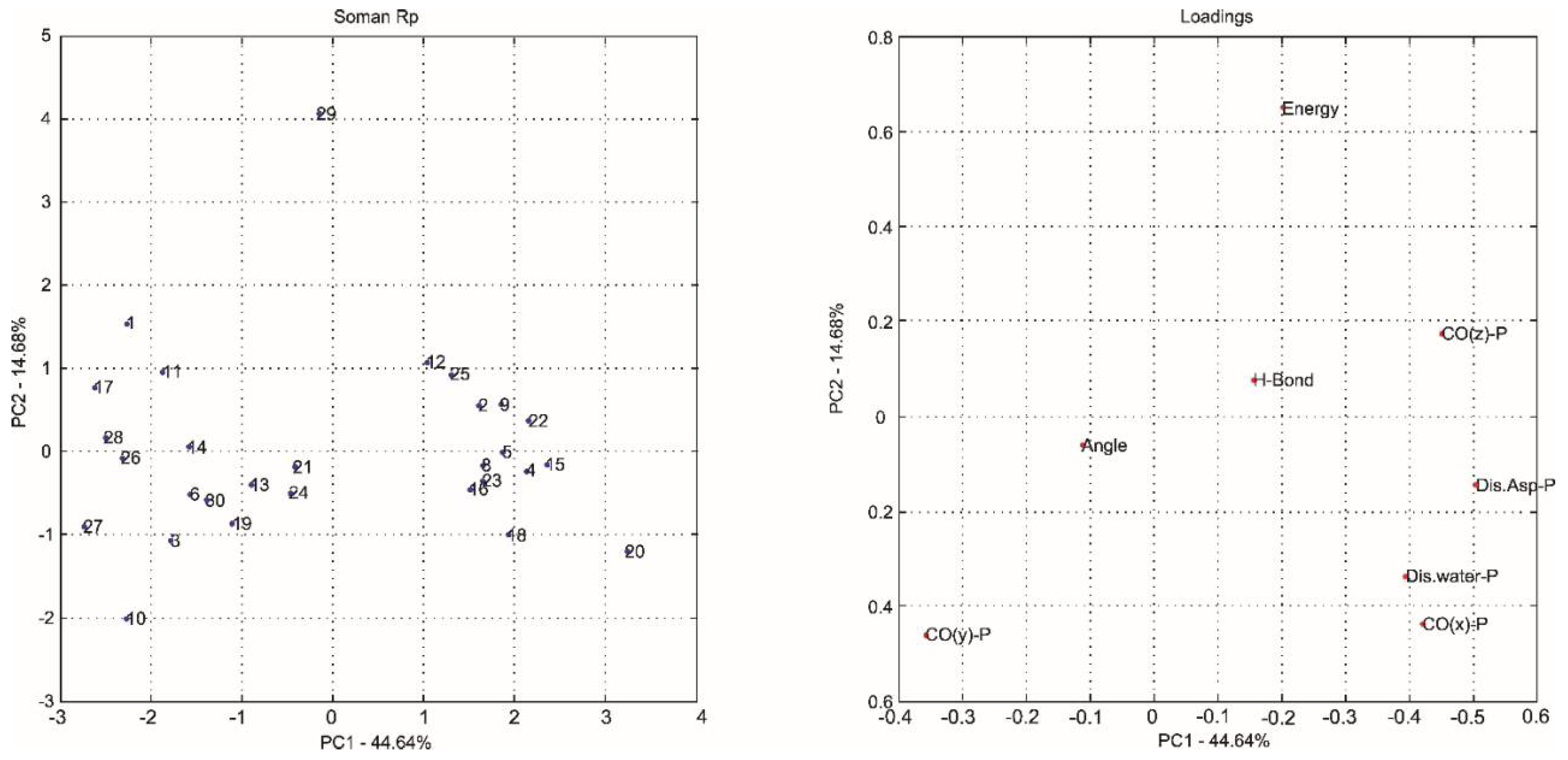
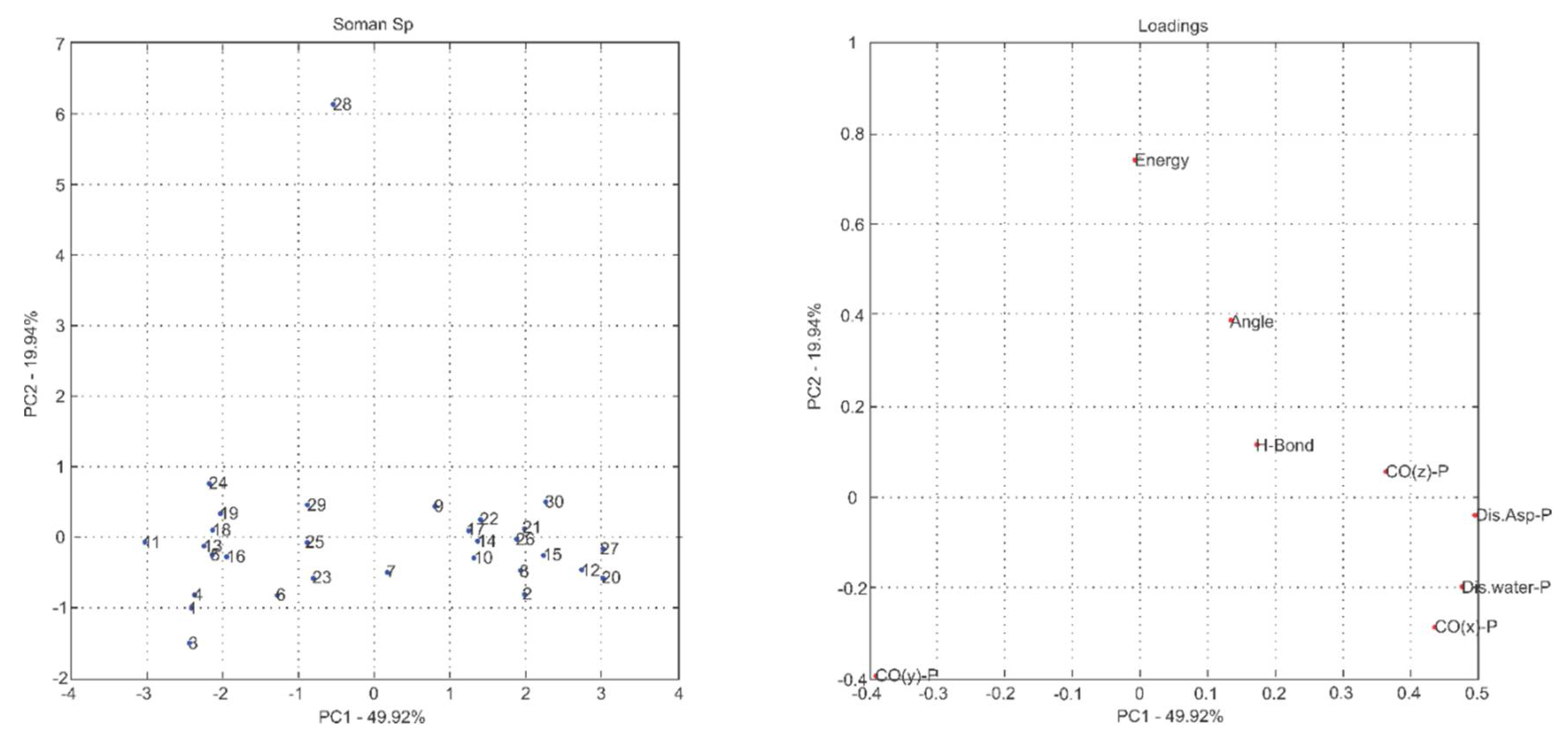
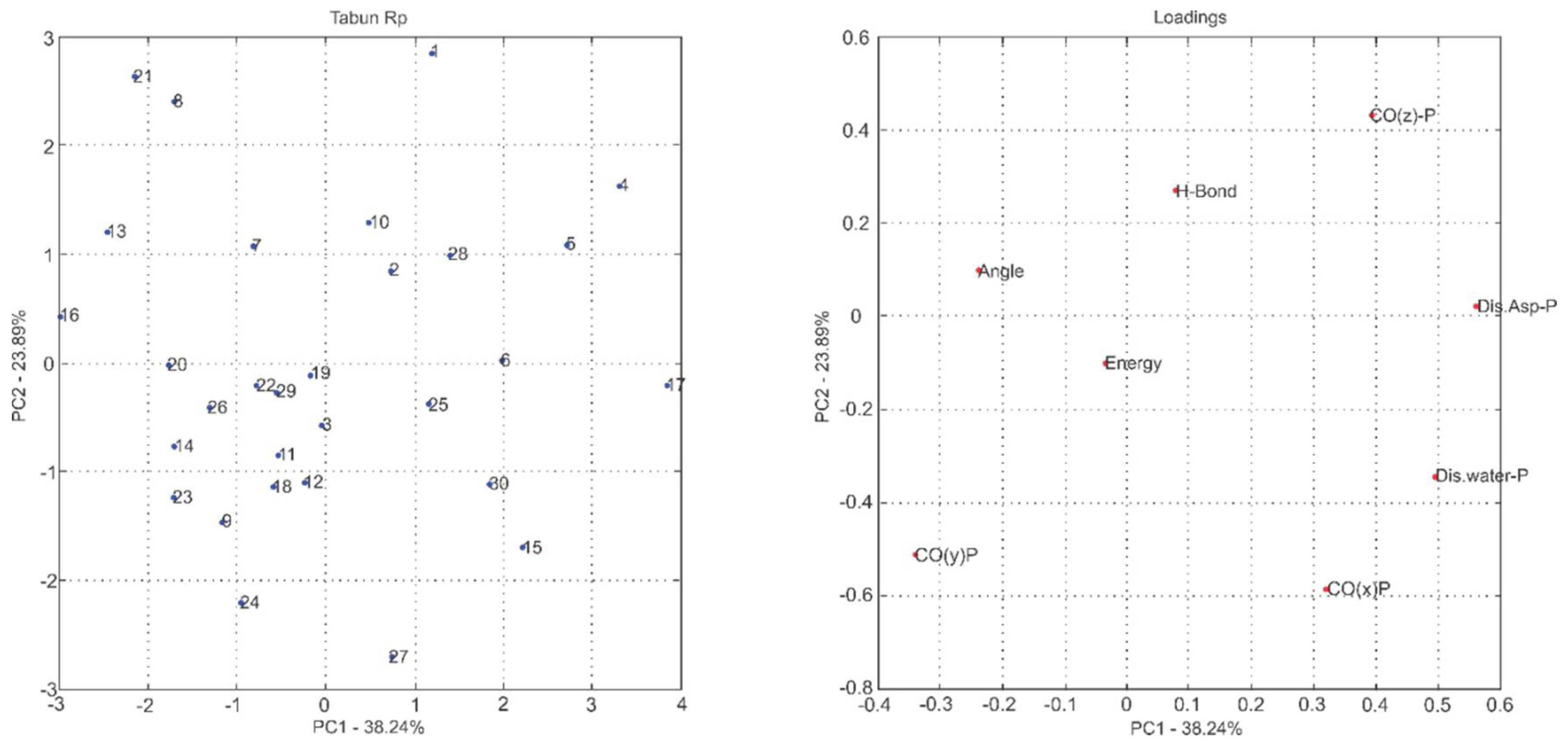
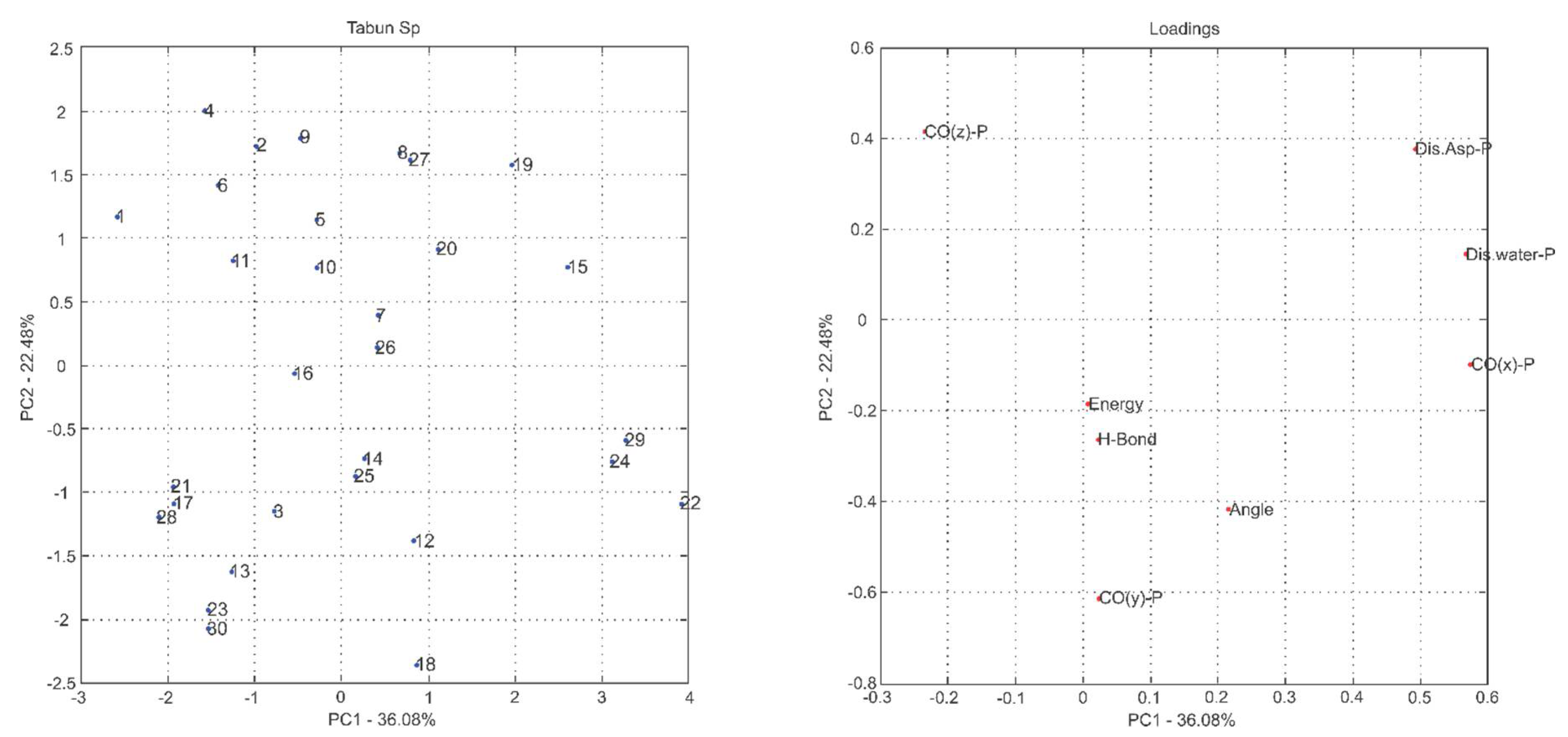
2.1. Multivariate Exploratory Analysis
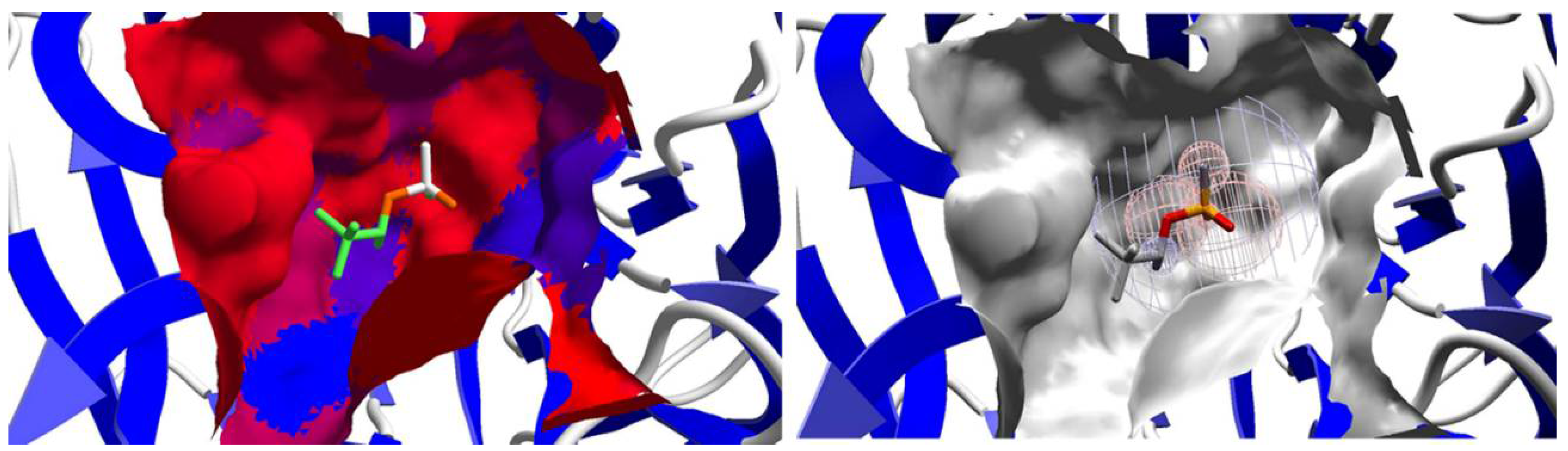
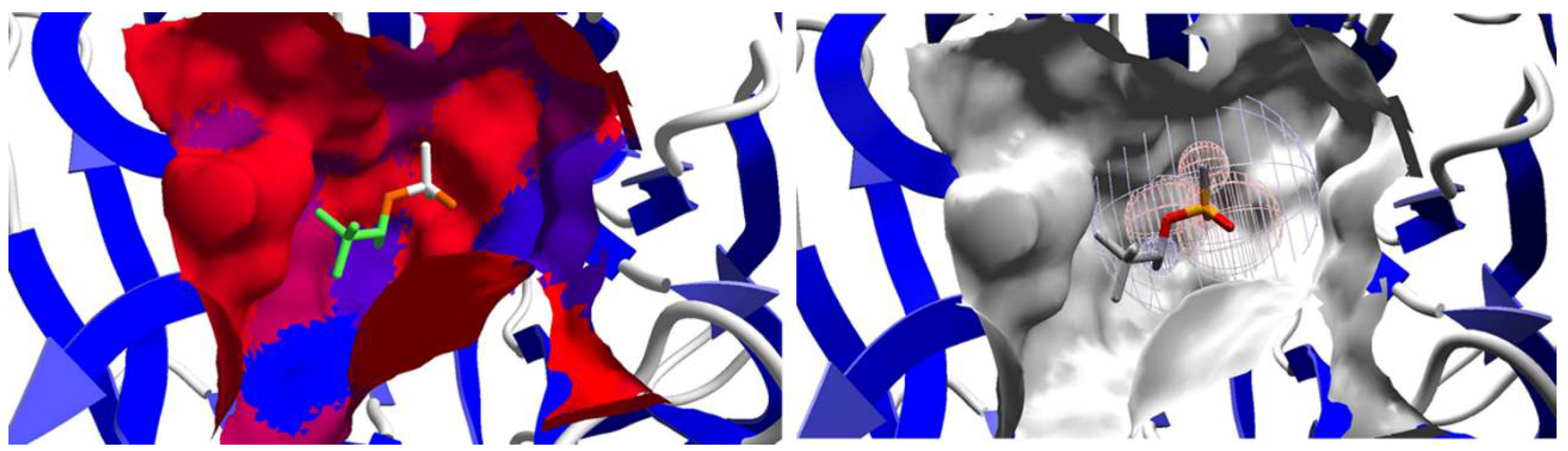
2.2. Affinity: Molecular Docking
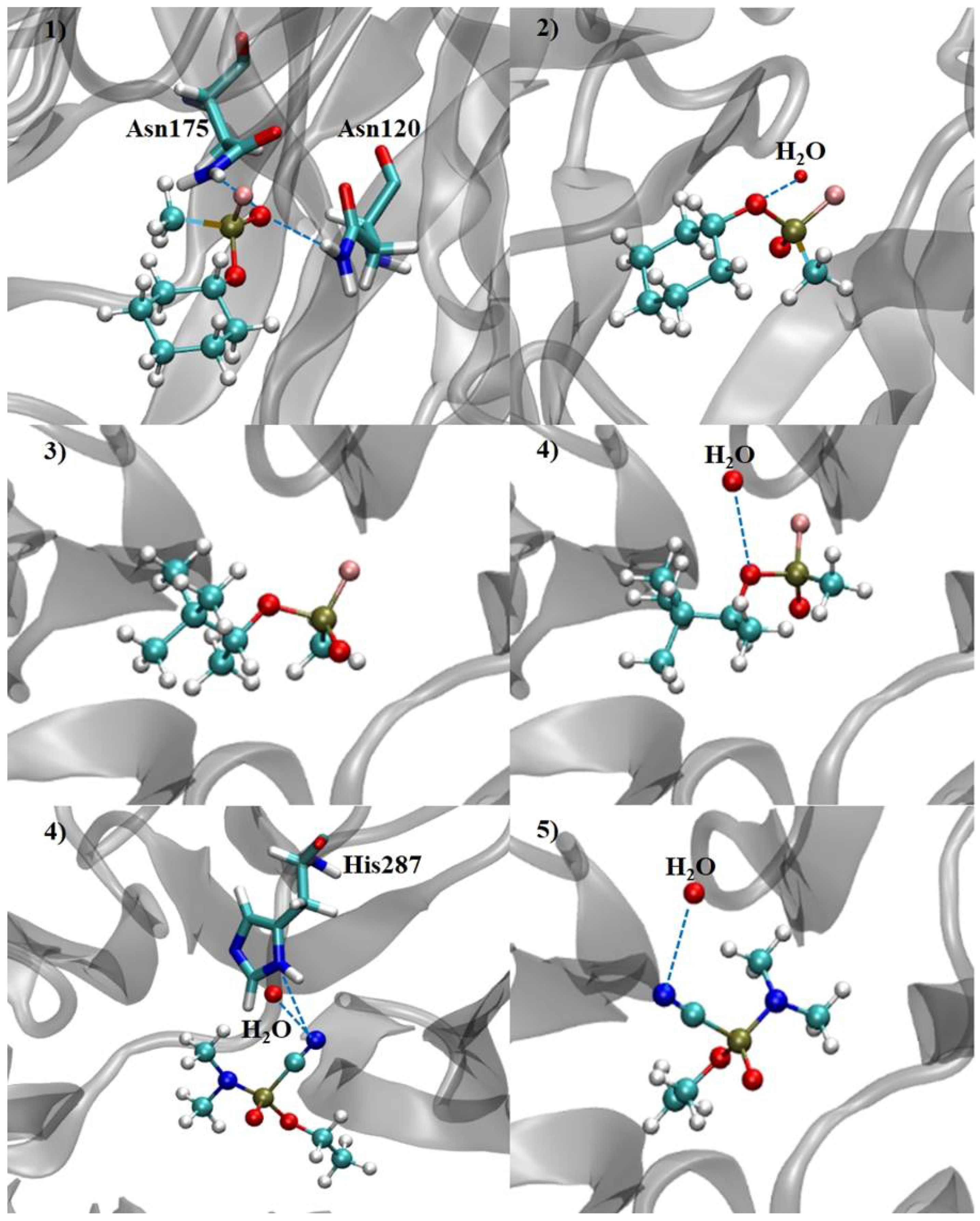
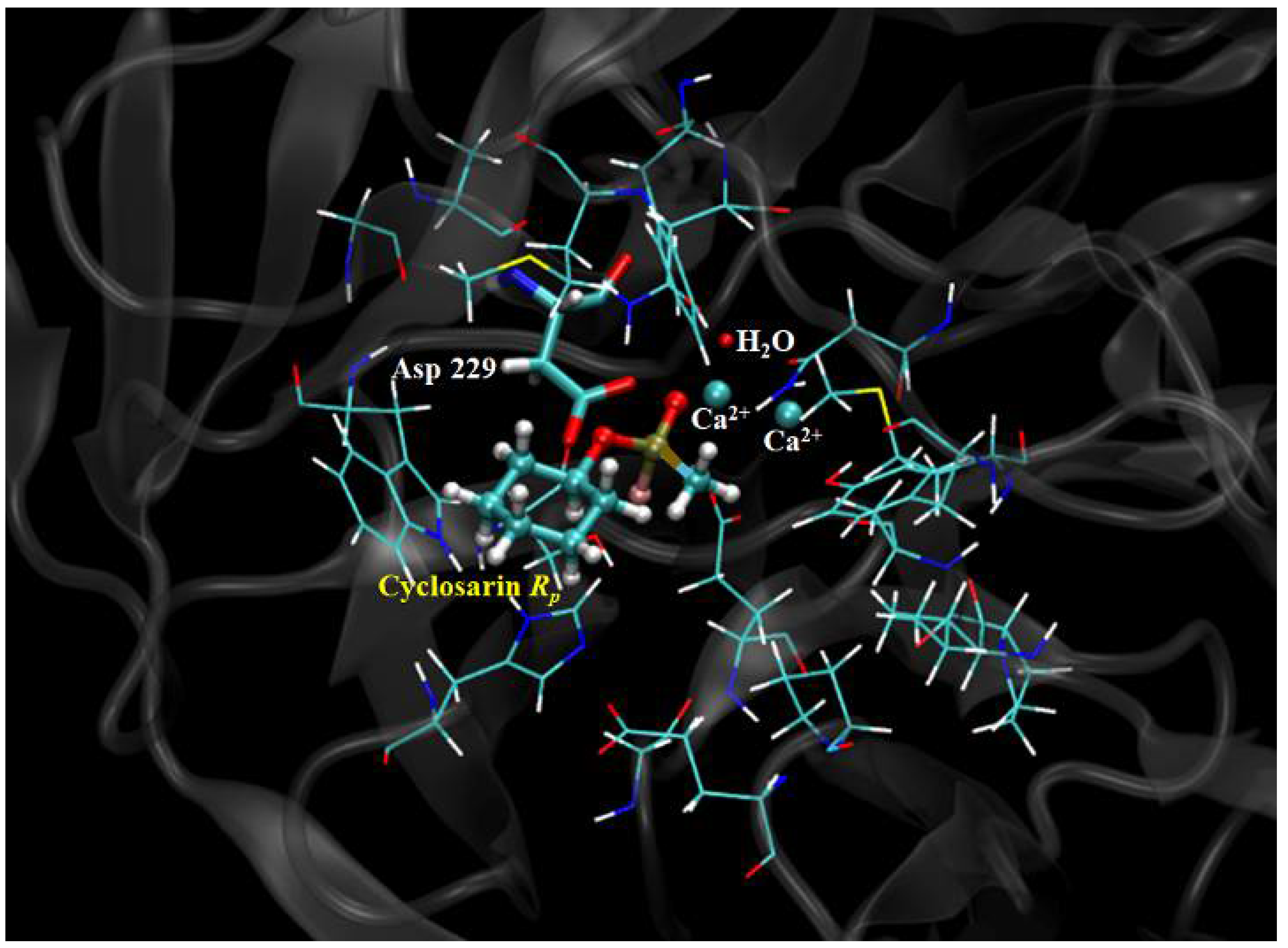
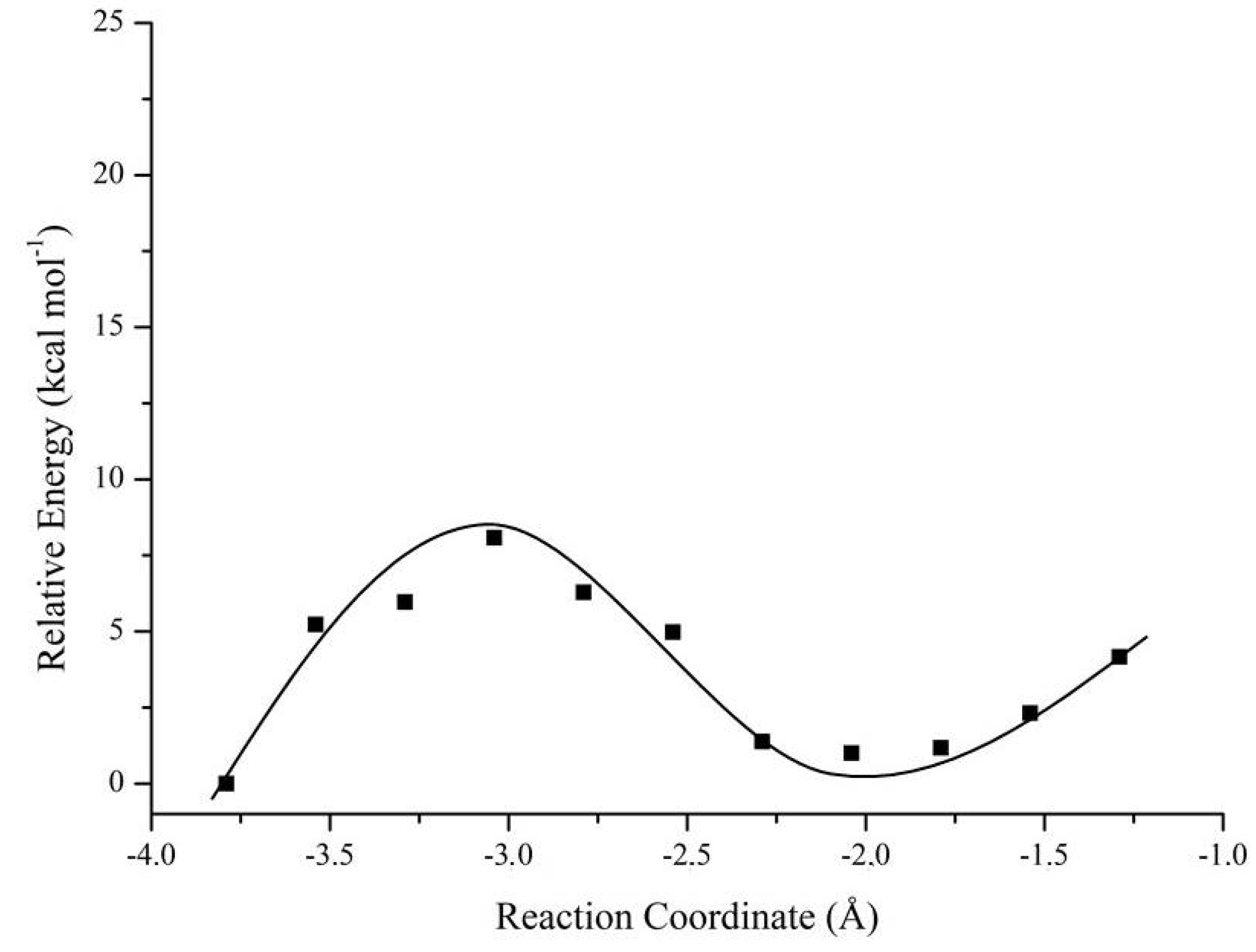
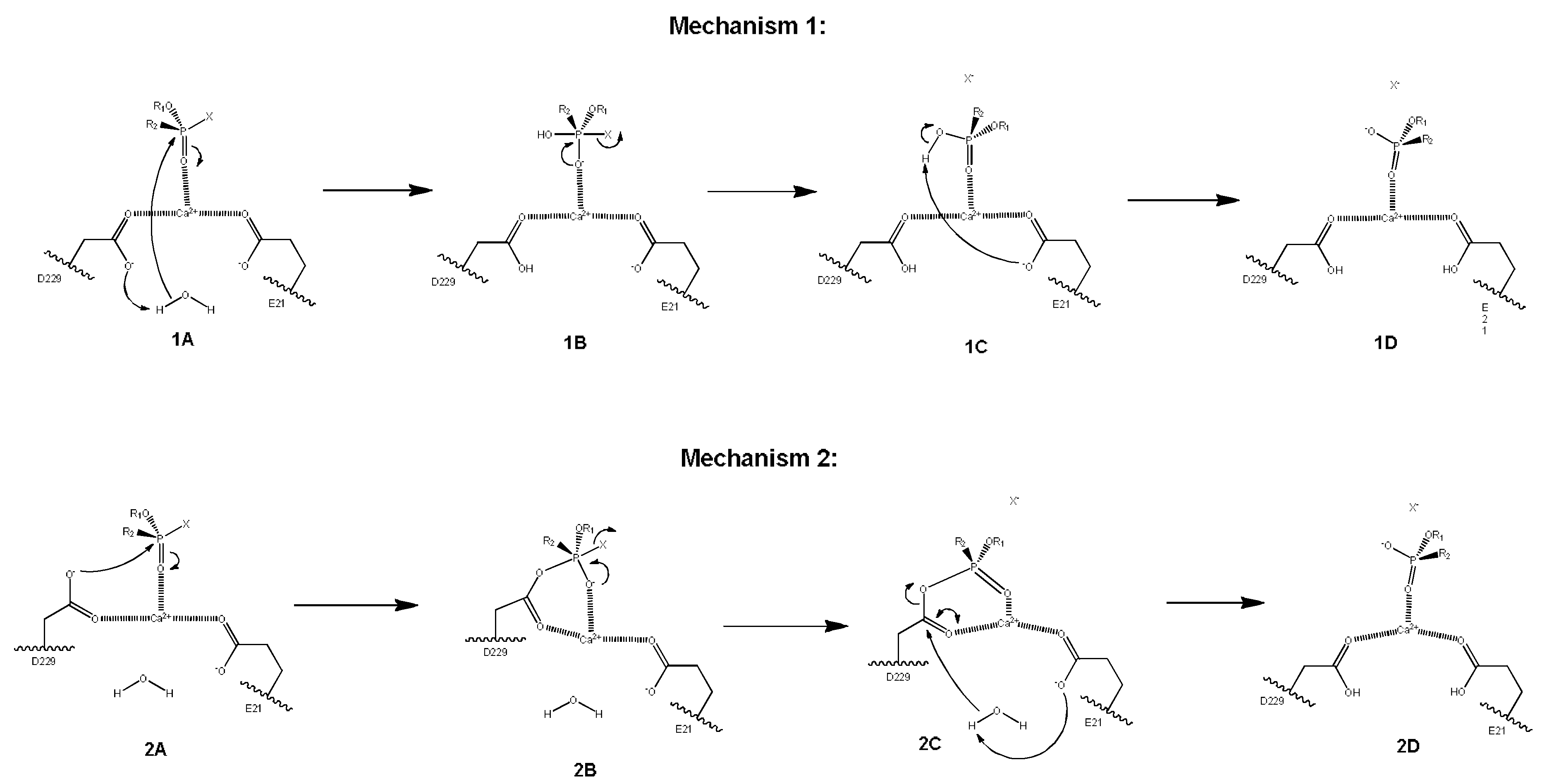
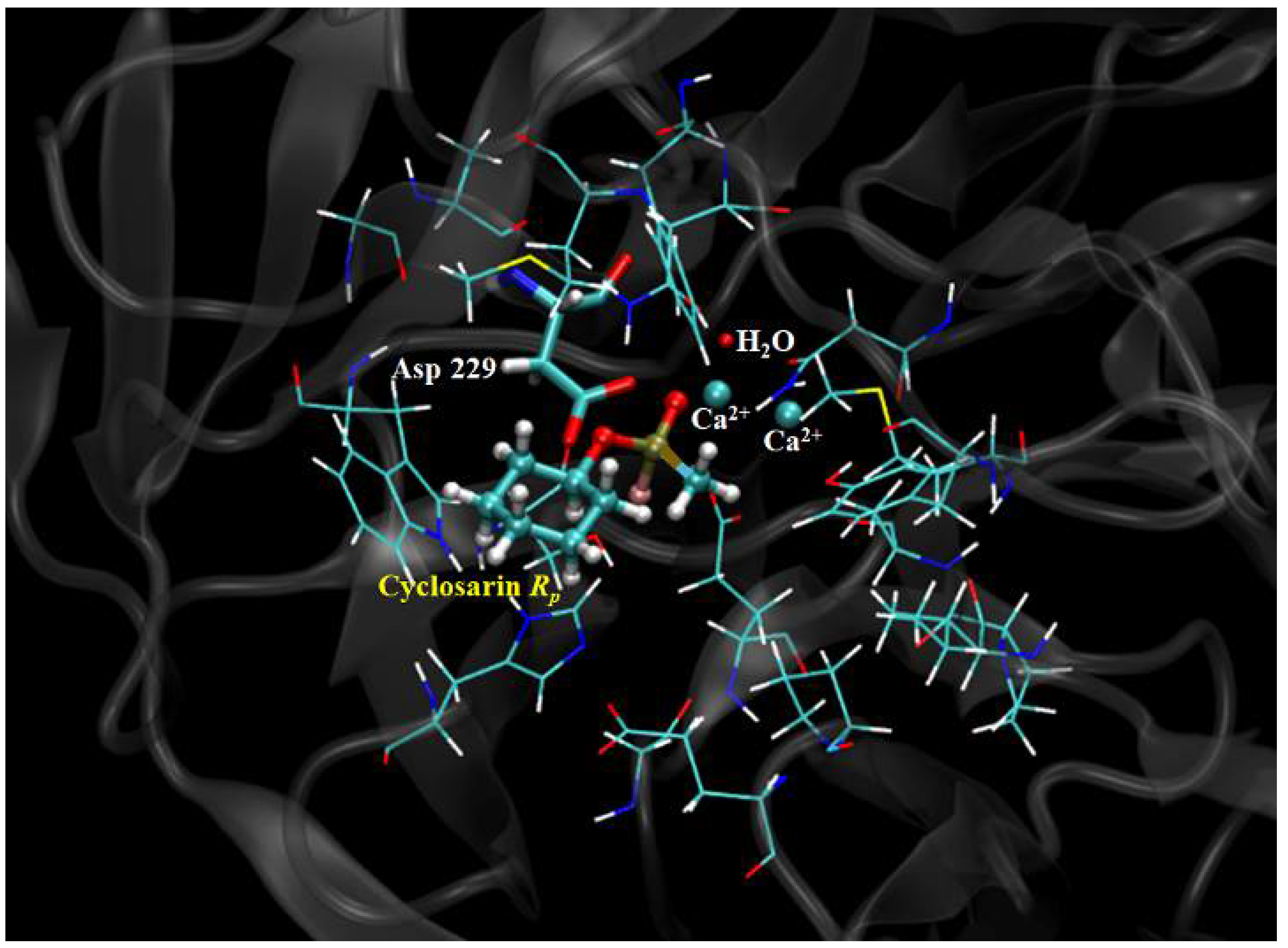
2.3. Mechanistic Studies in the Wild-Type DFPase Active Site
3. Materials and Methods
3.1. Computational Details
3.1.1. Docking Procedure
3.1.2. Multivariate Analysis of Principal Components
3.1.3. QM/MM Methodology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| Asn | Asparagine |
| Asp | Aspartate |
| B3LYP | DFT hybrid functional by Becke, Lee, Yang and Paar |
| DcPPA | Dicyclopentylphosphoroamidate |
| DFPase | Diisopropyl fluorophosphatase enzyme |
| DFT | Density Functional Theory |
| GA | NATO designation code for tabun |
| GB | NATO designation code for sarin |
| GD | NATO designation code for soman |
| GF | NATO designation code for cyclosarin |
| His | Histidine |
| Kcat | Turnover number |
| Km | Michaelis constant |
| M | Mols per liter |
| MM | Molecular Mechanics |
| MVD | Molegro Virtual Docker® software |
| OP | Organophosphorus |
| PC | Principal Component |
| PCA | Principal Components Analysis |
| PLP | Piecewise Linear Potential |
| QM | Quantum Mechanics |
| RP | R-stereochemistry designation for the OP phosphorus atom |
| SP | S-stereochemistry designation for the OP phosphorus atom |
References
- Chauhan, S.; Chauhan, S.; D’Cruz, R.; Faruqi, S.; Singh, K.K.; Varma, S.; Singh, M.; Karthik, V. Chemical warfare agents. Environ. Toxicol. Pharmacol. 2008, 26, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Carr, R.L. Biochemical mechanisms contributing to species differences in insecticidal toxicity. Toxicology 1995, 105, 291–304. [Google Scholar] [CrossRef]
- Li, J.N.; Liu, L.; Fu, Y.; Guo, Q.X. What are the pKa values of organophosphorus compounds? Tetrahedron 2006, 62, 4453–4462. [Google Scholar] [CrossRef]
- Jaga, K.; Dharmani, C. Sources of exposure to and public health implications of organophosphate pesticides. Rev. Panam. Salud Pública 2003, 14, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, J.O.S.; França, T.C.C.; Kuča, K.; Cunha, E.F.F.; Abagyan, R.; Mancini, D.T.; Ramalho, T.C. Molecular modeling and in vitro reactivation study between the oxime BI-6 and acetylcholinesterase inhibited by different nerve agents. J. Biomol. Struct. Dyn. 2015, 33, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.H.; Mustyakimov, M.; Schoenborn, B.P.; Langan, P.; Blum, M.M. Neutron structure and mechanistic studies of diisopropyl fluorophosphatase (DFPase). Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.; Chen, J.C.H.; Heidenreich, A.; Gäb, J.; Koller, M.; Kehe, K.; Blum, M.M. Reversed Enantioselectivity of Diisopropyl Fluorophosphatase against Organophosphorus Nerve Agents by Rational Design. J. Am. Chem. Soc. 2009, 131, 17226–17232. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.M. Commercializing chemical warfare: Citrus, cyanide, and an endless war. Agric. Hum. Values 2016, 33, 3–26. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Sod-Moriah, G.; Kaplan, D.; Mizrahi, D.; Segall, Y.; Kronman, C.; Karton, Y.; Lazar, A.; Marcus, D.; et al. Stereoselectivity toward VX is determined by interactions with residues of the acyl pocket as well as of the peripheral anionic site of AChE. Biochemistry 2004, 43, 11255–11265. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, T.C.; Castro, A.A.; Silva, D.R.; Silva, M.C.; França, T.C.C.; Bennion, B.J.; Kuca, K. Computational Enzymology and Organophosphorus Degrading Enzymes: Promising Approaches Toward Remediation Technologies of Warfare Agents and Pesticides. Curr. Med. Chem. 2016, 23, 1041–1061. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, L.; Yu, J.-G.; Liao, R.-Z. What roles do the residue Asp229 and the coordination variation of calcium play of the reaction mechanism of the diisopropyl-fluorophosphatase? A DFT investigation. Theor. Chem. Acc. 2016, 135, 138. [Google Scholar] [CrossRef]
- Wymore, T.; Field, M.J.; Langan, P.; Smith, J.C.; Parks, J.M. Hydrolysis of DFP and the Nerve Agent (S)-Sarin by DFPase Proceeds along Two Different Reaction Pathways: Implications for Engineering Bioscavengers. J. Phys. Chem. B 2014, 118, 4479–4489. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.M.; Löhr, F.; Richardt, A.; Ruterjans, H.; Chen, J.C.H. Binding of a Designed Substrate Analogue to Diisopropyl Fluorophosphatase: Implications for the Phosphotriesterase Mechanism. J. Am. Chem. Soc. 2006, 128, 12750–12757. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.A.; Caetano, M.S.; Silva, T.C.; Mancini, D.T.; Rocha, E.P.; Cunha, E.F.F.; Ramalho, T.C. Molecular Docking, Metal Substitution and Hydrolysis Reaction of Chiral Substrates of Phosphotriesterase. Comb. Chem. High Throughput Screen. 2016, 19, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, J.; Castro, A.A.; Ramalho, T.C.; Giacoppo, J.O.S.; Mancini, D.T.; Caetano, M.S.; Cunha, E.F.F. Asymmetric biocatalysis of the nerve agent VX by human serum paraoxonase 1: Molecular docking and reaction mechanism calculations. Med. Chem. Res. 2016, 25, 2521–2533. [Google Scholar] [CrossRef]
- Dawson, R.M.; Pantelidis, S.; Rose, H.R.; Kotsonis, S.E. Degradation of nerve agents by an organophosphate-degrading agent (OpdA). J. Hazard. Mater. 2008, 157, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Hanselman, D.; Littlefield, B. Mastering MATLAB 5: A Comprehensive Tutorial and Reference; Prentice-Hall: Bergen, NJ, USA, 1998; ISBN 978-0138583668. [Google Scholar]
- Lima, W.E.A.; Pereira, A.F.; Castro, A.A.; Cunha, E.F.F.; Ramalho, T.C. Flexibility in the Molecular Design of Acetylcholinesterase Reactivators: Probing Representative Conformations by Chemometric Techniques and Docking/QM Calculations. Lett. Drug Des. Discov. 2016, 13, 360–371. [Google Scholar] [CrossRef]
- Castro, A.A.; Assis, L.C.; Silva, D.R.; Corrêa, S.; Assis, T.M.; Gajo, G.C.; Soares, F.V.; Ramalho, T.C. Computational enzymology for degradation of chemical warfare agents: Promising technologies for remediation processes. AIMS Microbiol. 2017, 3, 108–135. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.C.; Pires, M.S.; Castro, A.A.; Cunha, E.F.F.; Caetano, M.S.; Ramalho, T.C. Molecular insight into the inhibition mechanism of plant and rat 4-hydroxyphenylpyruvate dioxygenase by molecular docking and DFT calculations. Med. Chem. Res. 2015, 24, 3958–3971. [Google Scholar] [CrossRef]
- Guimarães, A.P.; Oliveira, A.A.; Cunha, E.F.F.; Ramalho, T.C.; França, T.C.C. Analysis of Bacillus anthracis nucleoside hydrolase via in silico docking with inhibitors and molecular dynamics simulation. J. Mol. Model. 2011, 17, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Matos, K.S.; Mancini, D.T.; Cunha, E.F.F.; Kuca, K.; França, T.C.C.; Ramalho, T.C. Molecular aspects of the reactivation process of acetylcholinesterase inhibited by cyclosarin. J. Braz. Chem. Soc. 2011, 22, 1999–2004. [Google Scholar] [CrossRef]
- Ramalho, T.C.; Caetano, M.S.; Cunha, E.F.F.; Souza, T.C.S.; Rocha, M.V.J. Construction and Assessment of Reaction Models of Class I EPSP Synthase: Molecular Docking and Density Functional Theoretical Calculations. J. Biomol. Struct. Dyn. 2009, 27, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Cunha, E.F.F.; Barbosa, E.F.; Oliveira, A.A.; Ramalho, T.C. Molecular Modeling of Mycobacterium Tuberculosis DNA Gyrase and its Molecular Docking Study with Gatifloxacin Inhibitors. J. Biomol. Struct. Dyn. 2010, 27, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.C.S.; Josa, D.; Ramalho, T.C.; Caetano, M.S.; Cunha, E.F.F. Molecular modelling of Mycobacterium tuberculosis acetolactate synthase catalytic subunit and its molecular docking study with inhibitors. Mol. Simul. 2008, 34, 707–713. [Google Scholar] [CrossRef]
- Goncalves, A.S.; França, T.C.C.; Caetano, M.S.; Ramalho, T.C. Reactivation steps by 2-PAM of tabun-inhibited human acetylcholinesterase: Reducing the computational cost in hybrid QM/MM methods. J. Biomol. Struct. Dyn. 2014, 32, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Matos, K.S.; Cunha, E.F.F.; Abagyan, R.; Ramalho, T.C. Computational Evidence for the Reactivation Process of Human Acetylcholinesterase Inhibited by Carbamates. Comb. Chem. High Throughput Screen. 2014, 17, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, T.C.; Alencastro, R.B.; La-Scalea, M.A.; Figueroa-Villar, J.D. Theoretical evaluation of adiabatic and vertical electron affinity of some radiosensitizers in solution using FEP, ab initio and DFT methods. Biophys. Chem. 2004, 110, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Gustin, D.J.; Mattei, P.; Kast, P.; Wiest, O.; Lee, L.; Cleland, W.W.; Hilvert, D. Heavy Atom Isotope Effects Reveal a Highly Polarized Transition State for Chorismate Mutase. J. Am. Chem. Soc. 1999, 121, 1756–1757. [Google Scholar] [CrossRef]
- Giacoppo, J.O.S.; Mancini, D.T.; Guimarães, A.P.; Gonçalves, A.S.; Cunha, E.F.F.; França, T.C.C.; Ramalho, T.C. Molecular modeling toward selective inhibitors of dihydrofolate reductase from the biological warfare agent Bacillus anthracis. Eur. J. Med. Chem. 2015, 91, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, Y.; Zhang, J.; Chen, K.; Li, S.; Jiang, J. An isofenphos-methyl hydrolase (Imh) capable of hydrolyzing the P–O–Z moiety of organophosphorus pesticides containing an aryl or heterocyclic group. Appl. Microbiol. Biotechnol. 2012, 94, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Cunha, E.F.F.; Ramalho, T.C.; Reynolds, R.C. Binding Mode Analysis of 2,4-diamino-5-methyl-5-deaza-6-substituted Pteridines with Mycobacterium tuberculosis and Human Dihydrofolate Reductases. J. Biomol. Struct. Dyn. 2008, 25, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Van der Kamp, M.W.; Mulholland, A.J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Ranaghan, K.E.; Mulholland, A.J. Computational enzymology. Chem. Commun. 2010, 46, 2354–2372. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, K.; Mujika, J.I.; Ranaghan, K.E.; Manby, F.R.; Mulholland, A.J.; Harvey, J.N. Analysis of polarization in QM/MM modelling of biologically relevant hydrogen bonds. J. R. Soc. Interface 2008, 5, S207–S216. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Principal Components | % of Variance | % of Cumulative VARIANCE | Principal Components | % of Variance | % of Cumulative VARIANCE |
|---|---|---|---|---|---|
| (RP)-Cyclosarin | (SP)-Cyclosarin | ||||
| PC1 | 50.05 | 50.05 | PC1 | 47.55 | 47.55 |
| PC2 | 16.24 | 66.29 | PC2 | 21.32 | 68.87 |
| PC3 | 14.82 | 81.11 | PC3 | 12.88 | 81.75 |
| PC4 | 10.23 | 91.34 | PC4 | 8.98 | 90.73 |
| PC5 | 5.75 | 97.09 | PC5 | 5.98 | 96.71 |
| PC6 | 2.81 | 99.90 | PC6 | 3.21 | 99.92 |
| PC7 | 0.09 | 99.99 | PC7 | 0.07 | 99.99 |
| PC8 | 0.01 | 100.00 | PC8 | 0.01 | 100.00 |
| (RP)-Soman | (SP)-Soman | ||||
| PC1 | 44.64 | 44.64 | PC1 | 49.92 | 49.92 |
| PC2 | 14.68 | 59.32 | PC2 | 19.94 | 69.87 |
| PC3 | 13.48 | 72.80 | PC3 | 12.13 | 82.00 |
| PC4 | 11.63 | 84.43 | PC4 | 10.64 | 92.64 |
| PC5 | 6.83 | 91.26 | PC5 | 6.07 | 98.71 |
| PC6 | 5.25 | 96.51 | PC6 | 1.25 | 99.96 |
| PC7 | 3.43 | 99.94 | PC7 | 0.03 | 99.99 |
| PC8 | 0.06 | 100.00 | PC8 | 0.01 | 100.00 |
| (RP)-Tabun | (SP)-Tabun | ||||
| PC1 | 38.25 | 38.25 | PC1 | 36.08 | 36.08 |
| PC2 | 23.89 | 62.14 | PC2 | 22.48 | 58.56 |
| PC3 | 16.22 | 78.36 | PC3 | 15.66 | 74.22 |
| PC4 | 12.95 | 91.30 | PC4 | 11.39 | 85.61 |
| PC5 | 5.90 | 97.20 | PC5 | 8.83 | 94.44 |
| PC6 | 2.60 | 99.80 | PC6 | 5.34 | 99.78 |
| PC7 | 0.19 | 99.99 | PC7 | 0.13 | 99.91 |
| PC8 | 0.01 | 100.00 | PC8 | 0.09 | 100.00 |
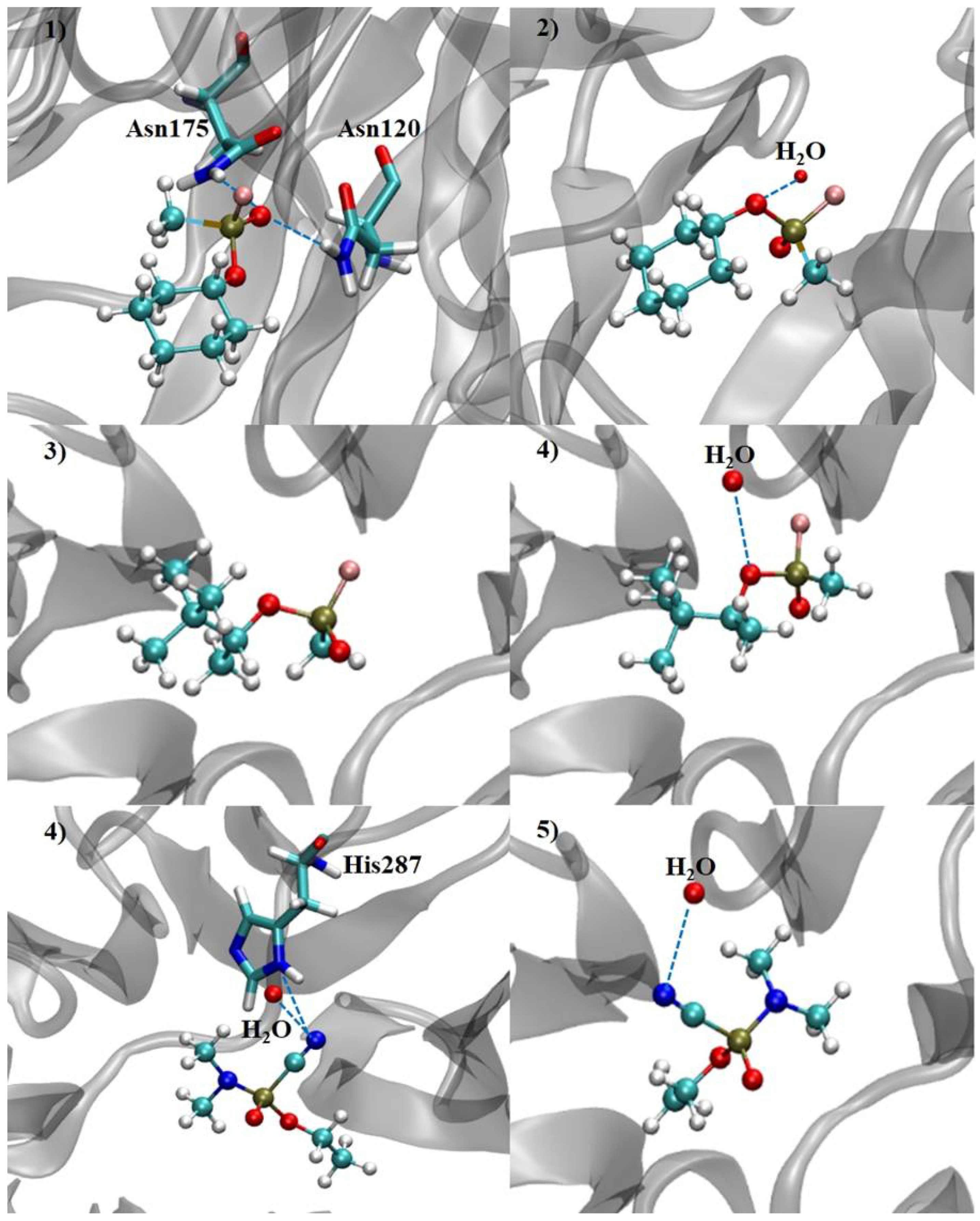
| Neurotoxic Agent | Intermolecular Interaction Energy | H-Bond Strength | H-Bond Length | Residues and H2O | Neurotoxic Agent | Intermolecular Interaction Energy | H-Bond Strength | H-Bond Length | Residues and H2O |
|---|---|---|---|---|---|---|---|---|---|
| (RP)-Cyclosarin | −39.23 | −1.58 | 2.99 | Asn120 | (SP)-Cyclosarin | −32.34 | −2.50 | 2.97 | H2O |
| −2.24 | 2.88 | Asn175 | |||||||
| (RP)-Soman | −40.88 | - | - | - | (SP)-Soman | −28.33 | −2.49 | 3.10 | H2O |
| (RP)-Tabun | −32.80 | −2.50 | 3.09 | H2O | (SP)-Tabun | −28.87 | −2.50 | 3.10 | H2O |
| −0.14 | 3.49 | His287 |
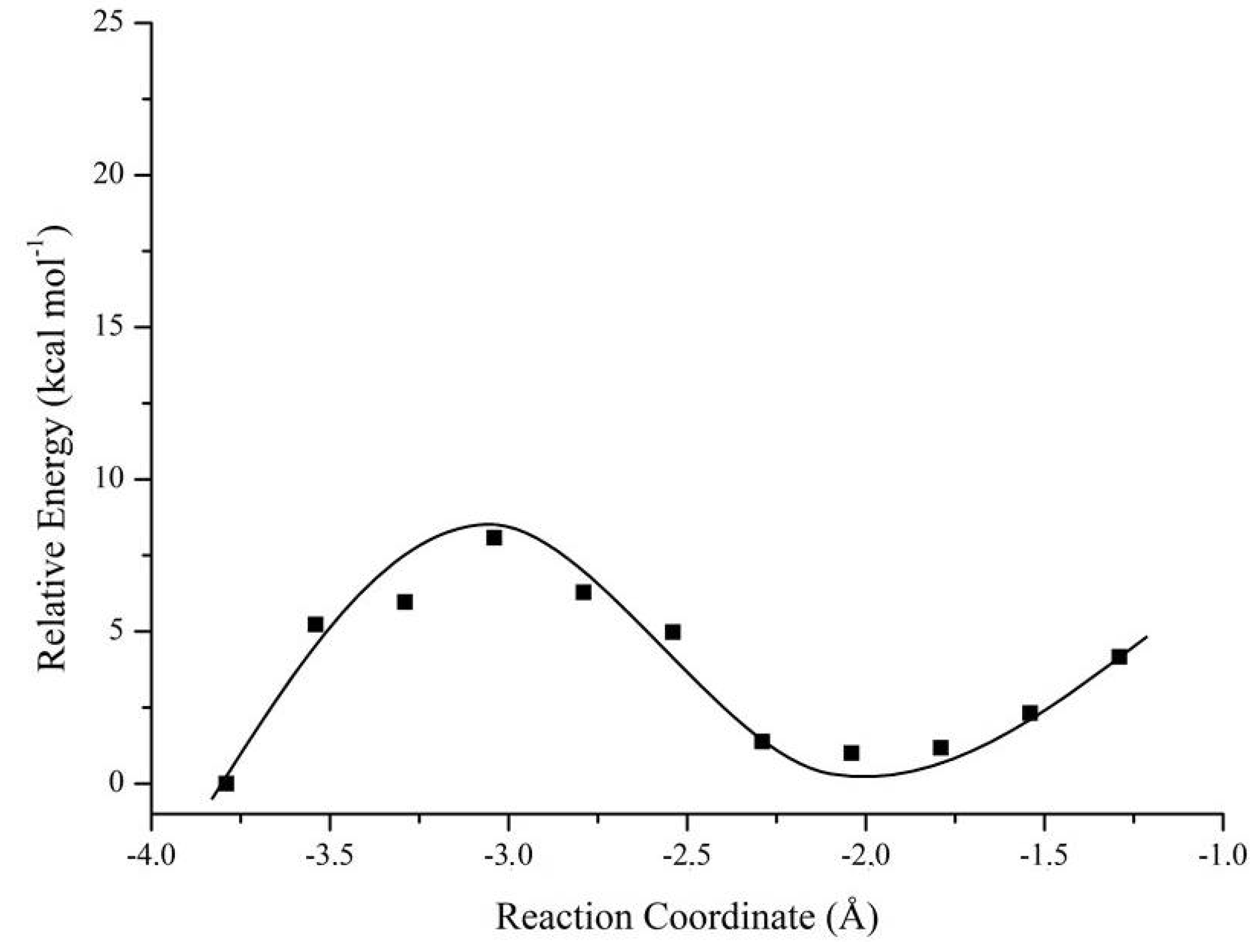
| Nerve Agent | Enantiomers | Mechanism 1; ΔΔE# (kcal·mol−1) | Mechanism 2; ΔΔE# (kcal·mol−1) |
|---|---|---|---|
| Soman | RP | 9.98 | 5.53 |
| SP | 0.00 | 1.81 | |
| Tabun | RP | 2.05 | 0.10 |
| SP | 2.21 | 2.52 | |
| Cyclosarin | RP | 2.11 | 1.68 |
| SP | 11.90 | 32.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, F.V.; De Castro, A.A.; Pereira, A.F.; Leal, D.H.S.; Mancini, D.T.; Krejcar, O.; Ramalho, T.C.; Da Cunha, E.F.F.; Kuca, K. Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication. Int. J. Mol. Sci. 2018, 19, 1257. https://doi.org/10.3390/ijms19041257
Soares FV, De Castro AA, Pereira AF, Leal DHS, Mancini DT, Krejcar O, Ramalho TC, Da Cunha EFF, Kuca K. Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication. International Journal of Molecular Sciences. 2018; 19(4):1257. https://doi.org/10.3390/ijms19041257
Chicago/Turabian StyleSoares, Flávia V., Alexandre A. De Castro, Ander F. Pereira, Daniel H. S. Leal, Daiana T. Mancini, Ondrej Krejcar, Teodorico C. Ramalho, Elaine F. F. Da Cunha, and Kamil Kuca. 2018. "Theoretical Studies Applied to the Evaluation of the DFPase Bioremediation Potential against Chemical Warfare Agents Intoxication" International Journal of Molecular Sciences 19, no. 4: 1257. https://doi.org/10.3390/ijms19041257