Scorpins in the DNA Damage Response


Department of Cancer Biology and Genetics, College of Medicine, The Ohio State University and James Comprehensive Cancer Center, Columbus, OH 43210, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(6), 1794; https://doi.org/10.3390/ijms19061794
Submission received: 21 May 2018
/
Revised: 11 June 2018
/
Accepted: 14 June 2018
/
Published: 17 June 2018
(This article belongs to the Special Issue Alterations to Signalling Pathways in Cancer Cells 2018)
Abstract
:The DNA Damage Response (DDR) is a complex signaling network that comes into play when cells experience genotoxic stress. Upon DNA damage, cellular signaling pathways are rewired to slow down cell cycle progression and allow recovery. However, when the damage is beyond repair, cells activate complex and still not fully understood mechanisms, leading to a complete proliferative arrest or cell death. Several conventional and novel anti-neoplastic treatments rely on causing DNA damage or on the inhibition of the DDR in cancer cells. However, the identification of molecular determinants directing cancer cells toward recovery or death upon DNA damage is still far from complete, and it is object of intense investigation. SPRY-containing RAN binding Proteins (Scorpins) RANBP9 and RANBP10 are evolutionarily conserved and ubiquitously expressed proteins whose biological functions are still debated. RANBP9 has been previously implicated in cell proliferation, survival, apoptosis and migration. Recent studies also showed that RANBP9 is involved in the Ataxia Telangiectasia Mutated (ATM) signaling upon DNA damage. Accordingly, cells lacking RANBP9 show increased sensitivity to genotoxic treatment. Although there is no published evidence, extensive protein similarities suggest that RANBP10 might have partially overlapping functions with RANBP9. Like RANBP9, RANBP10 bears sites putative target of PIK-kinases and high throughput studies found RANBP10 to be phosphorylated following genotoxic stress. Therefore, this second Scorpin might be another overlooked player of the DDR alone or in combination with RANBP9. This review focuses on the relatively unknown role played by RANBP9 and RANBP10 in responding to genotoxic stress.
1. Introduction
The DNA damage response (DDR) is an attractive target for anti-cancer treatments due to the increased genotoxic stress that malignant cells experience [1,2]. Modalities of repair of the damage are limited to a handful of recognized mechanisms (reviewed in [3]). In cancer cells, one or more of these mechanisms are deficient or altered, providing an opportunity of achieving synthetic lethality by inhibition of the remaining ones [4]. For example, BRCA-deficient malignancies are exquisitely sensitive to Poly(ADP)-Ribose Polymerase (PARP) inhibition [2,4,5]. Nonetheless, mounting evidence shows that cells adapt. They find ways to reactivate targeted/inactivated proteins or resort to alternative pathways guarantying DNA repair at levels compatible with survival and proliferation [2,5]. For these reasons, finding vulnerabilities that are specific to cancer cells sparing normal ones has proven to be challenging [2,5]. In addition, incomplete understanding of the network and missing tiles of the complex puzzle represented by unrecognized players are main reasons of the failure of anticancer treatments aiming to target DNA repair mechanisms [2,4,6].
SPRY-containing RAN binding Proteins (Scorpins) RANBP9 and RANBP10 are two highly similar proteins whose biological functions are poorly understood. This inadequate knowledge is due to multiple causes including the lack of validated antibodies able to discriminate between the two proteins. Moreover, the perinatal lethality of RANBP9 knockout mice has also negatively affected the investigation of its biological functions.
Comprehensive reviews about proposed biological roles of RANBP9 in cancer [7], Alzheimer’s disease and other contexts [8,9,10] are available. On the other hand, there are no exhaustive reviews about RANBP10 but only a few studies about its role in platelet physiology [11,12,13].
This review focuses on the relatively unknown role of Scorpins in responding to genotoxic stress.
2. Scorpins
RAN Binding Protein 9 (RANBP9) is a ~90 Kd scaffold protein that due to “a series of unfortunate events” has been labeled and probably investigated from the wrong perspective through the years. One of its first names was B-cell antigen receptor Ig Beta-Associated Protein 1, (IBAP-1) even if there is no evidence supporting any molecular role of this protein in B-cell signaling or the immune system in general (NCBI Gene ID: 56705). Another commonly used name is RANBPM (RAN Binding Protein Microtubule organizing center), which derives from the initial observation that the protein localized at the Microtubule Organizing Center (MTOC) [14]. However, a subsequent article from the same authors reported that they had been using only a truncated form of the wild type protein, lacking 229 amino acids at the N-terminus. Thus, they refuted their previous conclusion and showed that the full-length RANBP9 does not localize to the MTOC [15]. Finally, in the absence of a better functional characterization, the current preferred name of this scaffold protein is RAN Binding Protein 9 due to sequence homology with the RAN binding domain present on other proteins [14]. However, the evidence supporting a binding of RAN by RANBP9 is limited to the initial publication twenty years ago, was never reconfirmed and it is very weak at best [14]. For this reason, whether RANBP9 really binds and affects RAN functions remains to be formally proven and better investigated.
RAN Binding Protein 10 (RANBP10) was discovered as a paralog of RANBP9 and was given a name with the next numeral [16]. It has been shown in megakaryocytes that RANBP10 binds RAN and acts as a bona fide RAN-GTP Exchange Factor (GEF) in the cytoplasm although it showed a substantial lower specific activity than RCC1 (Regulator of Chromosome Condensation 1) RAN-GEF used as control in those experiments [11].
Because of their initial identification as RAN binding proteins, it was assumed that Scorpins participate in the nuclear-cytoplasmic import-export process [14,17]. In some databases RANBP9 is still confused with Importin 9, which is a distinct protein of 1048 amino acids (UniProtKB-Q96P70). While it is established that RANBP9 and RANBP10 are present both in the cell nucleus and cytoplasm, there is no evidence of a role of these two proteins in nuclear import/export. On the contrary, RANBP9 and RANBP10 are structurally very different from other RAN binding proteins (Figure 1). They bear four known domains and based on the distinctive presence of a PRY/SPRY domain they should more appropriately be called Scorpins, forming a separate subgroup by themselves as initially suggested by Hosono et al. [18]. For more information about the structure and the putative functions not related to the DDR of the different domains of Scorpins we recommend the reading of earlier reviews [7,8,10,19,20].
3. Known Biological Roles of RANBP9
RANBP9 is a ubiquitous scaffold protein present both in the cell nucleus and cytoplasm. It is evolutionarily highly conserved and its functions are poorly characterized [7,8]. The generation of RANBP9 knockout (KO) mice demonstrated that this protein is not required for embryonic development. However, most of RANBP9 KO animals die immediately after birth possibly because of lactation failure. The few survivors are smaller in size compared to their littermates and completely lack spermatogenesis or oogenesis [21]. Of note, RANBP9 has been linked to different types of cellular stress and might work as part of macromolecular complexes [7].
RANBP9 has been reported to directly or indirectly interact with more than a hundred proteins [7]. The Genecards database lists about 400 potential interactions (www.genecards.org). It is striking that the putative partners reported for RANBP9 do not share any significant structural homology or belong to one particular group of protein or cellular pathway [7,8]. On the contrary, RANBP9 putative interactors are very different among each other and participate in a wide variety of biological processes [7,8]. Many of the published interactions are derived from yeast-two-hybrid experiments or limited to co-immunoprecipitations performed upon exogenous over-expression of both RANBP9 and/or its putative partners. A good number of these interactions was never independently confirmed in subsequent studies and thus needs to be taken with caution. In several instances, the presence or absence of RANBP9 has been shown to have an effect on the amount of the interacting partner [22,23,24,25,26,27]. For example, RANBP9 has been shown to prevent the turnover of the mammalian homolog of the Lethal giant larvae (Mgl-1) tumor suppressor by facilitating the de-ubiquitination of the protein mediated by USP11 [23,28]. Also, relevant to the DDR, RANBP9 was shown to stabilize p73α [22]. However, how RANBP9 affects protein stability remains to be elucidated. In this regard, one potential mechanism of action in regulating protein turnover is through its role as corner stone necessary to assemble the CTLH complex [7,29,30], which is the mammalian equivalent of the Glucose-Induced degradation Deficient (GID) complex in yeast ([31,32] and Figure 2). The S. cerevisiae homolog of RANBP9 is GID1 and it is necessary for the formation of the entire complex, a putative E3 ligase macromolecular structure that degrades metabolic enzymes no longer needed after nutrient replenishment [31,32]. GID1 is also named VID30 (Vacuole-Induced degradation Deficient 30) because it can mediate the degradation of the same metabolic enzymes via lysosome [33,34]. Therefore, it is tempting to speculate that RANBP9 might regulate lysosome-mediated protein degradation. However, clear evidence supporting GID- or VID-like functions in mammalian cells is still lacking.
4. RANBP9 in the DDR
Several studies have linked RANBP9 to cellular response to multiple types of stress, including DNA damages upon exposure to ionizing radiation or chemotherapeutic drugs.
4.1. RANBP9 and Sensitivity to Ionizing Radiation (IR)
One of the most interesting unbiased studies was performed by Yard and colleagues, where 533 genetically annotated human cancer cell lines were analyzed by large scale profiling after exposure to ionizing radiation. This study identified RANBP9 as one of the top 19 genes associated with radiation sensitivity when mutated [35].
4.2. RANBP9 and Post-Translational Modifications Following Stress
Genotoxic stress caused by cisplatin, Ultra Violet (UV)-light, osmotic shock and Ionizing Radiation (IR) cell exposure [26,36] leads to RANBP9 phosphorylation by kinases that have been identified only to a limited extent. A list of predicted and experimentally proven Serine or Threonine phosphorylation target sites of PIK-kinases is shown in Table 1. However, other RANBP9 residues are predicted to be phosphorylated by other kinases that are not immediately relevant to the DDR and thus left out from our review. At least in part, specific post-translational modifications have been shown to influence both the sub-cellular localization of RANBP9 and its ability to bind different molecular partners, potentially mediating its biological functions [37].
4.3. RANBP9 Protein-Protein Interactions Relevant to the DDR
Several notable RANBP9 interactions reported in literature are with factors directly or indirectly associated to cellular DNA damage response [7]. These interactions might contribute to explain some of the phenotypes observed in the context of genotoxic stress when RANBP9 levels are altered.
Most of the reported RANBP9 interactions point to a potential involvement in determining apoptotic cell death by regulating mitochondrial function and membrane permeability, as described below. However, recent studies have demonstrated that many proposed RANBP9-interacting proteins in the nucleus can modulate cell ability to arrest cell cycle progression and affect repair of DNA upon different types of damages.
For example, RANBP9 over-expression has been shown to enhance dephosphorylation of the primary non-muscle isoform of ADF (Actin Depolymerizing Factor)/Cofilin (CFL-1) [38,39]. This event results in CFL-1 translocation to the mitochondria and promotes apoptosis [40,41]. However, it has been also demonstrated that overexpression of wild-type (phosphorylatable) CFL-1 can affect DNA repair capacity and results in cell death due to accumulation of damaged DNA. In fact, CFL-1 may localize into the nucleus preventing ATM-dependent phosphorylation of H2AX and recruitment of critical mediators of Homologous Dependent Repair (HDR) of DNA Double Strand Breaks (DSBs) [25,38,39,40,42,43,44,45]. Therefore, we can speculate that a decrease or complete ablation of RANBP9 might result in an increase of nuclear phosphorylated CFL1, which, in turn, causes a blunting of the ATM activation and DNA repair by HDR. Interestingly, the latter is in agreement with our group’s observations in lung cancer cells in which RANBP9 was silenced [26].
Wang and colleagues described the nuclear interaction between RANBP9 and Homeodomain Interacting Protein Kinase 2 (HIPK2) [46], a known mediator of apoptosis upon interaction with the death receptor Tumor Necrosis Factor (TNF) receptor type 1-associated death domain protein [46]. Notably, HIPK2 is activated upon phosphorylation by ATM and it controls DNA-damage dependent cell fate through the phosphorylation of key players of the DDR, such as p53 and the anti-apoptotic co-repressor C-terminal binding protein (CtBP) [47].
In a model of Alzheimer’s disease, Liu et al. demonstrated that RANBP9 physically associates with the tumor suppressor p73, enhancing its level both transcriptionally and post-transcriptionally, cooperating as a complex mediating mitochondria-dependent apoptosis [22,48]. Notably, p73 is a critical transcriptional regulator of genes involved in DNA repair, such as BRCA2, Rad51 and mre11 [49].
Of extreme interest from a DDR perspective is the reported interaction between Ubiquitin Specific Protease 11 (USP11) and RANBP9, which was considered responsible of the ubiquitination and degradation of the tumor suppressor Mgl1 [23,28]. However, recent studies have identified USP11-dependent p21 de-ubiquitination (which results in enhanced G2/M cell cycle checkpoint) [50] and regulation of the PALB2-BRCA1-BRCA2 interaction, required for the activation of HDR DNA repair specifically in the G2/M (and not in the G1) phase of the cell cycle [51]. Therefore, we can speculate that RANBP9 might influence USP11 functions in response to DNA damaging agents. It would be interesting to investigate whether RANBP9 nuclear accumulation during early phases of the DDR [26] has any effect on the PALB2-BRCA1-BRCA2 interaction.
Finally, both RANBP9 and RANBP10 interact with the Androgen Receptor (AR) and have been involved in regulation of AR-mediated transcription [52,53]. AR has been implicated in the DDR [54], particularly because of its ability to regulate the expression of multiple DNA repair genes (e.g., PARP1, FANCI, FANCC, MRE11A, ATR and NBN) involved in different steps of the DDR [55]. We can speculate that the nuclear accumulation during early phases of the DDR [26] might increase transcription of AR-dependent DDR genes. On the contrary, RANBP9 ablation might negatively impact this mechanism.
It is clear that a deeper investigation of RANBP9 interactions with recognized players of the DDR will be required to shed the light on novel mechanisms through which RANBP9 contributes to the response to genotoxic stress.
4.4. RANBP9 as a Target and Signaling Facilitator of the Ataxia Telangiectasia Mutated (ATM) Kinase
Our group has recently reported that, in response to IR, RANBP9 is phosphorylated on at least 3 different residues (S181, S550, and S603) by ATM, one of the most important kinases involved in the HDR of DNA DSBs. Upon ATM activation and phosphorylation, RANBP9 rapidly (2–8 h) accumulates in the nucleus and participates to the full activation of ATM, through a mechanism still not completely understood. Our results indicated that, in the absence of RANBP9, ATM acetylation, a post-translational modification linked to its complete activation [56,57,58,59,60], is reduced. This observation led us to hypothesize a model (Figure 3) in which, after DNA damage, RANBP9 is promptly recruited and accumulated into the nucleus upon ATM activation, to participate to its full activation, efficient DNA repair and cell survival. Conversely, when RANBP9 levels are abrogated, full activation of ATM and its downstream pathway are impaired, resulting in inefficient DNA repair and, eventually, cell death.
Several different questions remain open for this model, particularly how RANBP9 participates to ATM acetylation and full activation of ATM. A potential candidate for this molecular event is the histone acetyl-transferase (HAT) Tip60 (also know as KAT5), previously reported as RANBP9 interactor in the regulation of AICD transcriptional activity [61]. Interestingly, c-Abl-dependent Tip60 phosphorylation in response to DNA damage enhances its DNA binding activity and acetylation of ATM [56,57,58,59,60,62]. Further studies will be required to demonstrate the existence of a RANBP9/Tip60/ATM pathway required for the complete activation of DNA damage response.
Along the same lines, it is conceivable that, upon its quick nuclear accumulation, RANBP9 might attract into the nucleus additional factors directly or indirectly participating in repair of damaged DNA. However, since the protein is normally present in the nucleus, the currently available tools and reagents do not allow establishing whether RANBP9 is localized at foci of DNA damage.
4.5. RANBP9 as Pro-Apoptotic Tumor Suppressor
Our group has found that the ablation of RANBP9 in lung cancer cells renders them more sensitive to ionizing radiation and cisplatin exposure. In addition, when RANBP9 is silenced, the homology-directed DNA Repair mechanism is affected, dropping to about 50% of the controls cells [26]. However, earlier studies provide data that are in contrast with our findings [42,43,48,63]. For example, the Kang’s lab has shown that RANBP9 cooperates with p73 to induce apoptosis. Over-expression of RANBP9 in neuronal cells alters the Bax/Bcl2 ratio, promotes Bax oligomerization and induces Cytochrome C from the mitochondria. Simultaneously, mitochondrial fragmentation is observed [42,43,48]. These observations were made in neuronal cells without testing the sensitivity to DNA damaging agents.
The Schild-Poulter lab has provided additional evidence for a pro-apoptotic tumor suppressive role of RANBP9 [63]. Using HeLa cells, they have shown that upon exposure to ionizing radiation RANBP9 accumulates into the cytoplasm at times longer than 24 hours. However, they did not study the dynamics of RANBP9 accumulation at earlier time points. In addition to showing that over-expression of RANBP9 causes cell death, they also showed that HeLa cells in which RANBP9 was silenced are less sensitive to ionizing radiation in direct contrast with our observations. In addition, they show that when RANBP9 was absent levels of Bcl2 are increased in the cytoplasm and less Bax is present at the mitochondria [63].
While it is conceivable that the different cellular models and conditions might explain the contrasting results, it is also important to note that RANBP9 (and RANBP10) are factors involved in maintaining cellular homeostasis and working in concert with other proteins. Therefore, some of the results obtained by forced over-expression or silencing of RANBP9 might have the same deleterious end-result on cellular fitness.
In summary, based on a growing body of evidence, it is conceivable that RANBP9 exerts multifaceted effects in DDR depending on multiple factors like the type of genotoxic stress, the cellular context, and its molecular sub-cellular localization.
5. Known Biological Roles of RANBP10
RANBP10 is the least studied of the two Scorpins. The “lack of interest” in this protein is at least in part due to the absence of an obvious phenotype of the RANBP10 knockout mouse. In fact, genetic inactivation of RANBP10 in mouse does not cause early post-natal lethality or infertility like reported for RANBP9 [21,64]. Specialized studies established that RANBP10 knockout mice have a mild platelet deficit due to microtubule functional anomalies [13,65]. Furthermore, RANBP10 is expressed at lower levels compared to RANBP9 and it was shown that RANBP10 not only was not able to increase activation of the RAS/ERK pathway like its paralog, but was competing for the binding of cMET at the membrane and inhibiting the kinase signaling mediated by RANBP9 [16]. However, in addition to the significant structural similarities and the overlapping ubiquitous expression with RANBP9, RANBP10 was pulled-down as component of the CTLH complex similarly to its paralog ([29,30] and Figure 2C). Furthermore, the two proteins were co-immunoprecipitated and shown to have an overlapping cellular distribution by immunofluorescence [53]. In the Genecards database and excluding the interaction with RANBP9, there is currently a list of 87 proteins putatively interacting with RANBP10. Only 15 of those potential interactions are not shared with its paralog. Therefore, it is conceivable that RANBP9 and RANBP10 might have overlapping functions and is able to compensate for each other in physiological conditions or in their collective roles in the DDR.
6. RANBP10 in the DDR
Based on the hypothesis that Scorpins may have overlapping functions, the role of RANBP10 in the DDR needs to be elucidated. Up-to-date, there are no studies linking directly RANBP10 to the DDR or showing that RANBP10 ablation has an effect on the DDR signaling network as we have shown for RANBP9. An analysis of the RANBP10 amino acidic sequence reveals the presence of several sites putative target of PIK kinases (Table 1). In particular, S69 within the PRY domain is included in a long stretch (51 amino acids) that is highly similar to the region on RANBP9, where it corresponds to ATM-phosphorylated S181. We have previously demonstrated that ATM phosphorylates RANBP9 S181 in vitro using a peptide that is present in both Scorpins ([26] and Table 1). Therefore, we can speculate that RANBP10 S69 is a direct target of ATM too.
RANBP10 was not identified as a target of ATM or ATR in the seminal study by Matsuoka et al., in which they found RANBP9 S603 phosphorylated by ATM [66]. However, subsequent high throughput studies comparing proteomics before and after genotoxic stress list RANBP10 as protein subject to post-translational modifications. For example, S361, S365, and S369 are phosphorylated following ionizing radiation. The same data set reports only RANBP9 S477 as phosphorylated [67]. In Pines et al. (2011), residues S363, S389, S397 are reported as phosphorylated [68]. In Elia et al. (2015), RANBP10 K293 is acetylated following UV [69]. While these reported post-translational modification following genotoxic stress does not constitute compelling evidence, they nevertheless suggest that RANBP10 is involved in the DDR. Also, some of these reported sites are not predicted by our analysis in Table 1. Therefore, future in depth investigations will be required to establish their relevance in the context of the DDR.
7. Future Perspectives
Scorpins are poorly studied in general. Therefore, there are many open questions that need to be answered to clarify their biological roles and their involvement in the DDR specifically. One immediate question that can be addressed and prioritized is whether the ablation of RANBP10 alone or in combination with RANBP9 affects the sensitivity to genotoxic stress. Experiments inducing DNA damage in RANBP10 KO cells or mice are not reported. Due to the clinical implications, it is important to establish if the absence of RANBP10 sensitize cancer cells to ionizing radiation or genotoxic drugs in general in a similar manner to what we reported for RANBP9. We do not yet know whether RANBP10 behave similarly in cells following DNA damage. Most importantly, it is necessary to elucidate what are the molecular mechanisms through which the Scorpins contribute to the DDR. As of now, we still do not know how RANBP9 and RANBP10 protect cells from genotoxic stress. In response to DNA damage, Scorpins may act in the context of the CTLH complex or independently. The involvement of the CTLH complex raises the question whether Scorpins determine protein turnover of important players of the DNA repair.
On a more basic level, the clinical relevance of Scorpins expression/protein levels still requires extensive studies. The lack of validated reagents, particularly anti-RANBP10 specific antibodies, has severely limited the investigation of the relevance of these proteins in the clinical setting. For example, it is not yet possible to establish how well the levels of mRNA correlate with the Scorpin protein amounts. The generation of cell and mouse models by the use of the latest technologies will be necessary to answer many of the open questions. Likewise, a holistic proteomics approach will be essential in establishing how Scorpins through their multiple interactions coordinate the intricate cellular networks responsible for DNA repair, cell cycle arrest, disposal of potentially damaged organelles or protein aggregates upon DDR activation and cell death.
Finally, since Scorpins are involved in responding to different types of stress, from a clinical perspective, it would be interesting whether their absence can sensitize tumors to hyperthermia, for example. In fact, hyperthermia has been shown to sensitize tumors to genotoxic treatments and several DDR players are sensitive to temperature increases [70,71,72].
In summary, the investigation on the role of Scorpins in the cellular DDR is only at initial phases and it is necessary to establish the molecular details of RANBP9 and RANBP10 in determining the most appropriate response to damage of the DNA and whether they can be considered as a viable target for cancer treatment.
Funding
A.T. is recipient of a Pelotonia Fellowship for Cancer Research. This work was supported by start-up funds to V.C. from the College of Medicine, Office of Research, and Comprehensive Cancer Center of the Ohio State University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair. (Amst) 2018, 66–67, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Vanhecke, E.; Olaussen, K.A.; Lord, C.J.; Ashworth, A.; Soria, J.C. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat. Rev. Clin. oncol. 2012, 9, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Helena, J.M.; Joubert, A.M.; Grobbelaar, S.; Nolte, E.M.; Nel, M.; Pepper, M.S.; Coetzee, M.; Mercier, A.E. Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, L.M.; Maitland, M.E.R.; McTavish, C.J.; Schild-Poulter, C. Cell signalling pathway regulation by RanBPM: Molecular insights and disease implications. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Ramakrishna, S.; Baek, K.H. Diverse roles of the scaffolding protein RanBPM. Drug Discov. Today 2012, 17, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Puverel, S.; Tessarollo, L. RanBPM, a scaffolding protein for gametogenesis. Curr. Top. Dev. Biol. 2013, 102, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Murrin, L.C.; Talbot, J.N. RanBPM, a scaffolding protein in the immune and nervous systems. J. Neuroimmune Pharmacol. 2007, 2, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Dose, M.; Korpal, M.; Meyer, I.; Italiano, J.E., Jr.; Shivdasani, R.A. RanBP10 is a cytoplasmic guanine nucleotide exchange factor that modulates noncentrosomal microtubules. J. Biol. Chem. 2008, 283, 14109–14119. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R. Getting in shape with RanBP10. Blood 2009, 114, 5412–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, I.; Kunert, S.; Schwiebert, S.; Hagedorn, I.; Italiano, J.E., Jr.; Dutting, S.; Nieswandt, B.; Bachmann, S.; Schulze, H. Altered microtubule equilibrium and impaired thrombus stability in mice lacking RanBP10. Blood 2012, 120, 3594–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Masuda, H.; Horii, J.; Kuma, K.; Yokoyama, N.; Ohba, T.; Nishitani, H.; Miyata, T.; Tanaka, M.; Nishimoto, T. When overexpressed, a novel centrosomal protein, RanBPM, causes ectopic microtubule nucleation similar to gamma-tubulin. J. Cell Biol. 1998, 143, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Hirose, E.; Uchimura, Y.; Nakamura, M.; Umeda, M.; Nishii, K.; Mori, N.; Nishimoto, T. Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene 2001, 272, 25–33. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Schoen, S.R.; Messing, E.M.; Wu, G. A novel MET-interacting protein shares high sequence similarity with RanBPM, but fails to stimulate MET-induced Ras/Erk signaling. Biochem. Biophys. Res. Commun. 2004, 313, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T. A new role of ran GTPase. Biochem. Biophys. Res. Commun. 1999, 262, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Noda, S.; Shimizu, A.; Nakanishi, N.; Ohtsubo, M.; Shimizu, N.; Minoshima, S. YPEL5 protein of the YPEL gene family is involved in the cell cycle progression by interacting with two distinct proteins RanBPM and RanBP10. Genomics 2010, 96, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Suresh, B.; Kim, H.H.; Ramakrishna, S. RanBPM: A potential therapeutic target for modulating diverse physiological disorders. Drug Discov. Today 2017, 22, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Haq, S.; Ramakrishna, S. Scaffolding protein RanBPM and its interactions in diverse signaling pathways in health and disease. Discov. Med. 2018, 25, 177–194. [Google Scholar] [PubMed]
- Puverel, S.; Barrick, C.; Dolci, S.; Coppola, V.; Tessarollo, L. RanBPM is essential for mouse spermatogenesis and oogenesis. Development 2011, 138, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.; Ozaki, T.; Miyazaki, K.; Kato, C.; Hanamoto, T.; Nakagawara, A. Protein stability and function of p73 are modulated by a physical interaction with RanBPM in mammalian cultured cells. Oncogene 2005, 24, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Ramakrishna, S.; Kim, Y.S.; Kim, S.M.; Kim, M.S.; Baek, K.H. Stability and function of mammalian lethal giant larvae-1 oncoprotein are regulated by the scaffolding protein RanBPM. J. Biol. Chem. 2010, 285, 35340–35349. [Google Scholar] [CrossRef] [PubMed]
- Atabakhsh, E.; Schild-Poulter, C. RanBPM is an inhibitor of ERK signaling. PLoS ONE 2012, 7, e47803. [Google Scholar] [CrossRef] [PubMed]
- Palavicini, J.P.; Wang, H.; Minond, D.; Bianchi, E.; Xu, S.; Lakshmana, M.K. RanBP9 overexpression down-regulates phospho-cofilin, causes early synaptic deficits and impaired learning, and accelerates accumulation of amyloid plaques in the mouse brain. J. Alzheimers Dis. 2014, 39, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Scarpa, M.; Tessari, A.; Uka, R.; Amari, F.; Lee, C.; Richmond, T.; Foray, C.; Sheetz, T.; Braddom, A.; et al. Ran Binding Protein 9 (RanBP9) is a novel mediator of cellular DNA damage response in lung cancer cells. Oncotarget 2016, 7, 18371–18383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puverel, S.; Kiris, E.; Singh, S.; Klarmann, K.D.; Coppola, V.; Keller, J.R.; Tessarollo, L. RanBPM (RanBP9) regulates mouse c-Kit receptor level and is essential for normal development of bone marrow progenitor cells. Oncotarget 2016, 7, 85109–85123. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Suresh, B.; Park, J.H.; Kim, Y.S.; Ramakrishna, S.; Baek, K.H. Ubiquitin-specific protease 11 functions as a tumor suppressor by modulating Mgl-1 protein to regulate cancer cell growth. Oncotarget 2016, 7, 14441–14457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Texier, Y.; Toedt, G.; Gorza, M.; Mans, D.A.; van Reeuwijk, J.; Horn, N.; Willer, J.; Katsanis, N.; Roepman, R.; Gibson, T.J.; et al. Elution profile analysis of SDS-induced subcomplexes by quantitative mass spectrometry. Mol. Cell. Proteomics 2014, 13, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Menssen, R.; Schweiggert, J.; Schreiner, J.; Kusevic, D.; Reuther, J.; Braun, B.; Wolf, D.H. Exploring the topology of the Gid complex, the E3 ubiquitin ligase involved in catabolite-induced degradation of gluconeogenic enzymes. J. Biol. Chem. 2012, 287, 25602–25614. [Google Scholar] [CrossRef] [PubMed]
- Santt, O.; Pfirrmann, T.; Braun, B.; Juretschke, J.; Kimmig, P.; Scheel, H.; Hofmann, K.; Thumm, M.; Wolf, D.H. The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol. Biol. Cell 2008, 19, 3323–3333. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.R.; McCann, J.A.; Chiang, H.L. The heat shock protein Ssa2p is required for import of fructose-1, 6-bisphosphatase into Vid vesicles. J. Cell Biol. 2000, 150, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Alibhoy, A.A.; Giardina, B.J.; Dunton, D.D.; Chiang, H.L. Vid30 is required for the association of Vid vesicles and actin patches in the vacuole import and degradation pathway. Autophagy 2012, 8, 29–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yard, B.D.; Adams, D.J.; Chie, E.K.; Tamayo, P.; Battaglia, J.S.; Gopal, P.; Rogacki, K.; Pearson, B.E.; Phillips, J.; Raymond, D.P.; et al. A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat. Commun. 2016, 7, 11428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denti, S.; Sirri, A.; Cheli, A.; Rogge, L.; Innamorati, G.; Putignano, S.; Fabbri, M.; Pardi, R.; Bianchi, E. RanBPM is a phosphoprotein that associates with the plasma membrane and interacts with the integrin LFA-1. J. Biol. Chem. 2004, 279, 13027–13034. [Google Scholar] [CrossRef] [PubMed]
- Salemi, L.M.; Loureiro, S.O.; Schild-Poulter, C. Characterization of RanBPM molecular determinants that control its subcellular localization. PLoS ONE 2015, 10, e0117655. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.D.; Chiu, Y.W.; Lo, C.C.; Chiang, P.H.; Chiu, S.J.; Tsai, C.H.; Hwang, J.J.; Chen, R.C.; Gorbunova, V.; Lee, Y.J. Enhanced cellular radiosensitivity induced by cofilin-1 over-expression is associated with reduced DNA repair capacity. Int. J. Radiat. Biol. 2013, 89, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Leu, J.D.; Lee, Y.J. The actin depolymerizing factor (ADF)/cofilin signaling pathway and DNA damage responses in cancer. Int. J. Mol. Sci. 2015, 16, 4095–4120. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Boggess, T.; Uhlar, C.; Wang, X.; Khan, H.; Cappos, G.; Joly-Amado, A.; De Narvaez, E.; Majid, S.; Minamide, L.S.; et al. RanBP9 at the intersection between cofilin and Abeta pathologies: Rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 2015, 6, 1676. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013, 25, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.E.; Woo, J.A.; Lakshmana, M.K.; Uhlar, C.; Ankala, V.; Boggess, T.; Liu, T.; Hong, Y.H.; Mook-Jung, I.; Kim, S.J.; et al. Mitochondrial dysfunction and calcium deregulation by the RanBP9-cofilin pathway. FASEB J. 2013, 27, 4776–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.A.; Jung, A.R.; Lakshmana, M.K.; Bedrossian, A.; Lim, Y.; Bu, J.H.; Park, S.A.; Koo, E.H.; Mook-Jung, I.; Kang, D.E. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ. 2012, 19, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, X.; Hu, G.; Li, X.; Zhuang, Z.; Liu, J.; Wu, D.; Yang, L.; Xu, X.; Huang, X.; et al. Poly(ADP-ribose) glycohydrolase silencing down-regulates TCTP and Cofilin-1 associated with metastasis in benzo(a)pyrene carcinogenesis. Am. J. Cancer Res. 2015, 5, 155–167. [Google Scholar] [PubMed]
- Lee, Y.J.; Sheu, T.J.; Keng, P.C. Enhancement of radiosensitivity in H1299 cancer cells by actin-associated protein cofilin. Biochem. Biophys. Res. Commun. 2005, 335, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Marion Schneider, E.; Li, X.; Duttenhofer, I.; Debatin, K.; Hug, H. HIPK2 associates with RanBPM. Biochem. Biophys. Res. Commun. 2002, 297, 148–153. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Glas, C.; Bitomsky, N. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays 2013, 35, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Roh, S.E.; Woo, J.A.; Ryu, H.; Kang, D.E. Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death Dis. 2013, 4, e476. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Sengupta, S.; Gurdziel, K.; Bell, G.W.; Jacks, T.; Flores, E.R. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009, 5, e1000680. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Yan, G.; Song, X.; Xie, L.; Zhou, Y.; Li, J.; Hu, X.; Li, Z.; Hu, J.; Zhang, Y.; et al. Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proc. Natl. Acad. Sci. USA 2018, 115, 4678–4683. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.A.; Cheng, H.; Quayle, A.N.; Nishitani, H.; Nelson, C.C.; Rennie, P.S. RanBPM, a nuclear protein that interacts with and regulates transcriptional activity of androgen receptor and glucocorticoid receptor. J. Biol. Chem. 2002, 277, 48020–48027. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Yokoyama, T.; Yamaji, R.; Nakano, Y.; Inui, H. RanBP10 acts as a novel coactivator for the androgen receptor. Biochem. Biophys. Res. Commun. 2008, 368, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.C.; Li, L. Connecting androgen receptor signaling and the DNA damage response: Development of new therapies for advanced prostate cancer. Mol. Cell. Oncol. 2017, 4, e1321167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Adamowicz, M.; Vermezovic, J.; d'Adda di Fagagna, F. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016, 16, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Singha, N.; O'Connell, M.J.; Ronai, Z.A. Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 2008, 283, 17605–17614. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Claverie, P.; Salon, C.; Leduc, C.; Col, E.; Brambilla, E.; Khochbin, S.; Gazzeri, S. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol. Cell. Biol. 2006, 26, 4339–4350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, S.C.; Konietzko, U.; Henriques, A.G.; Rebelo, S.; Fardilha, M.; Nishitani, H.; Nitsch, R.M.; da Cruz, E.S.E.F.; da Cruz, E.S.O.A. RanBP9 modulates AICD localization and transcriptional activity via direct interaction with Tip60. J. Alzheimers Dis. 2014, 42, 1415–1433. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Jackson, S.P. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 2013, 498, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atabakhsh, E.; Bryce, D.M.; Lefebvre, K.J.; Schild-Poulter, C. RanBPM has proapoptotic activities that regulate cell death pathways in response to DNA damage. Mol. Cancer Res. 2009, 7, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Palavicini, J.P.; Lloyd, B.N.; Hayes, C.D.; Bianchi, E.; Kang, D.E.; Dawson-Scully, K.; Lakshmana, M.K. RanBP9 Plays a Critical Role in Neonatal Brain Development in Mice. PLoS ONE 2013, 8, e66908. [Google Scholar] [CrossRef] [PubMed]
- Kunert, S.; Meyer, I.; Fleischhauer, S.; Wannack, M.; Fiedler, J.; Shivdasani, R.A.; Schulze, H. The microtubule modulator RanBP10 plays a critical role in regulation of platelet discoid shape and degranulation. Blood 2009, 114, 5532–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Pines, A.; Kelstrup, C.D.; Vrouwe, M.G.; Puigvert, J.C.; Typas, D.; Misovic, B.; de Groot, A.; von Stechow, L.; van de Water, B.; Danen, E.H.; et al. Global phosphoproteome profiling reveals unanticipated networks responsive to cisplatin treatment of embryonic stem cells. Mol. Cell. Biol. 2011, 31, 4964–4977. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.; Crezee, J.; Franken, N.A.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat. Oncol. 2015, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- van Oorschot, B.; Granata, G.; Di Franco, S.; Ten Cate, R.; Rodermond, H.M.; Todaro, M.; Medema, J.P.; Franken, N.A. Targeting DNA double strand break repair with hyperthermia and DNA-PKcs inhibition to enhance the effect of radiation treatment. Oncotarget 2016, 7, 65504–65513. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.; Caspari, T. When heat casts a spell on the DNA damage checkpoints. Open Biol. 2014, 4, 140008. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of RANBP9 and RANBP10 proteins. RANBP10 shares high amino acid conservation with RANBP9 in the PRY (94%), SPRY (97%), LisH (82%), CTLH (90%), and CRA (89%) domains. The two proteins differ the most at the N-terminus and in the post-CTLH region, which contains several putative PIK-kinase phosphorylation sites (see Table 1). PRD (red)= Proline-Rich Domain; PRY/SPRY (light blue) = Spore lysis A and Ryanodine receptor Domain; LisH (green) = Lissencephaly type-I-like homology motif; CTLH (yellow) = Carboxy-terminal to LisH motif domain; CRA (orange) = CT11-RanBP9 domain; dark blue = putative Nuclear Localization Signal; Tub (purple) = tubulin-binding domain.
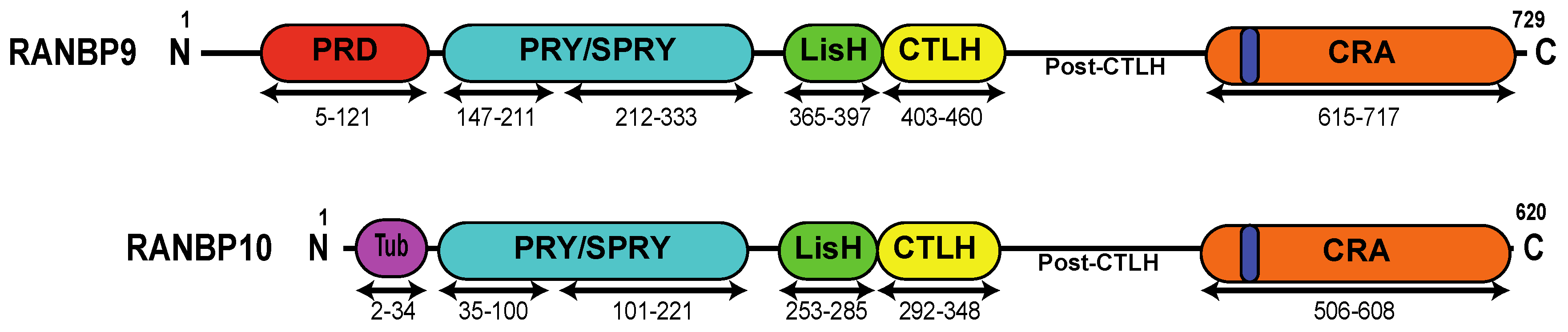
Figure 1.
Schematic representation of RANBP9 and RANBP10 proteins. RANBP10 shares high amino acid conservation with RANBP9 in the PRY (94%), SPRY (97%), LisH (82%), CTLH (90%), and CRA (89%) domains. The two proteins differ the most at the N-terminus and in the post-CTLH region, which contains several putative PIK-kinase phosphorylation sites (see Table 1). PRD (red)= Proline-Rich Domain; PRY/SPRY (light blue) = Spore lysis A and Ryanodine receptor Domain; LisH (green) = Lissencephaly type-I-like homology motif; CTLH (yellow) = Carboxy-terminal to LisH motif domain; CRA (orange) = CT11-RanBP9 domain; dark blue = putative Nuclear Localization Signal; Tub (purple) = tubulin-binding domain.
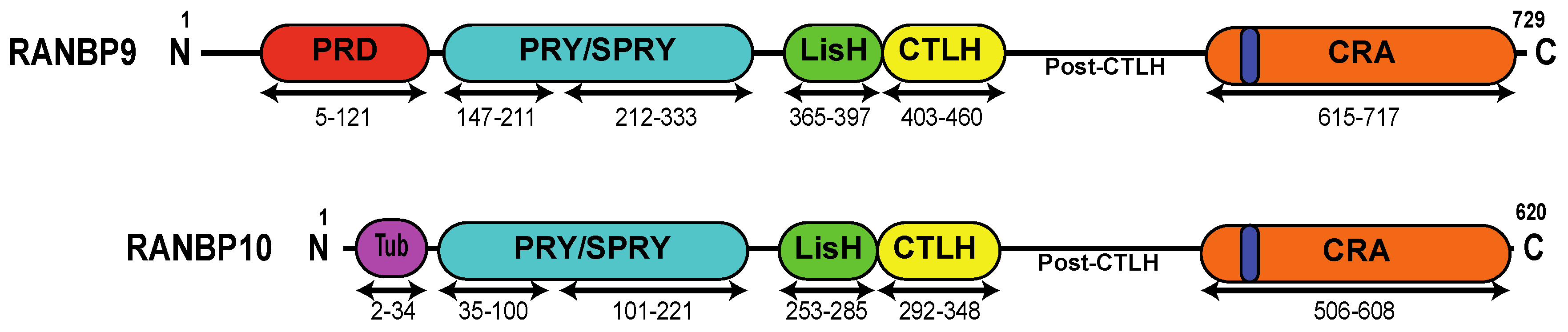
Figure 2.
Schematic representation of the S. cerevisiae Glucose-Induced degradation Deficient (GID)- and correspondent mammalian CTLH-macromolecular complexes. (A) The topology of the GID complex in yeast is well established [31]; (B) Predicted composition of the mammalian CTLH complex based on the GID mammalian homologs. The name of the complex comes from the CTLH domain that most of the members have; (C) CTLH or Nuclear Receptor coregulator-complex pulled down from mammalian cells including RANBP9 and RANBP10 (adapted from [29]). In the depicted complex, GID8 is named C20orf11 and indicated as C20. The experiment showed as part of the complex also YPEL5 (Yippee Like 5) indicated as Y5 in the cartoon, which has no known equivalent in the S. cerevisiae GID complex.
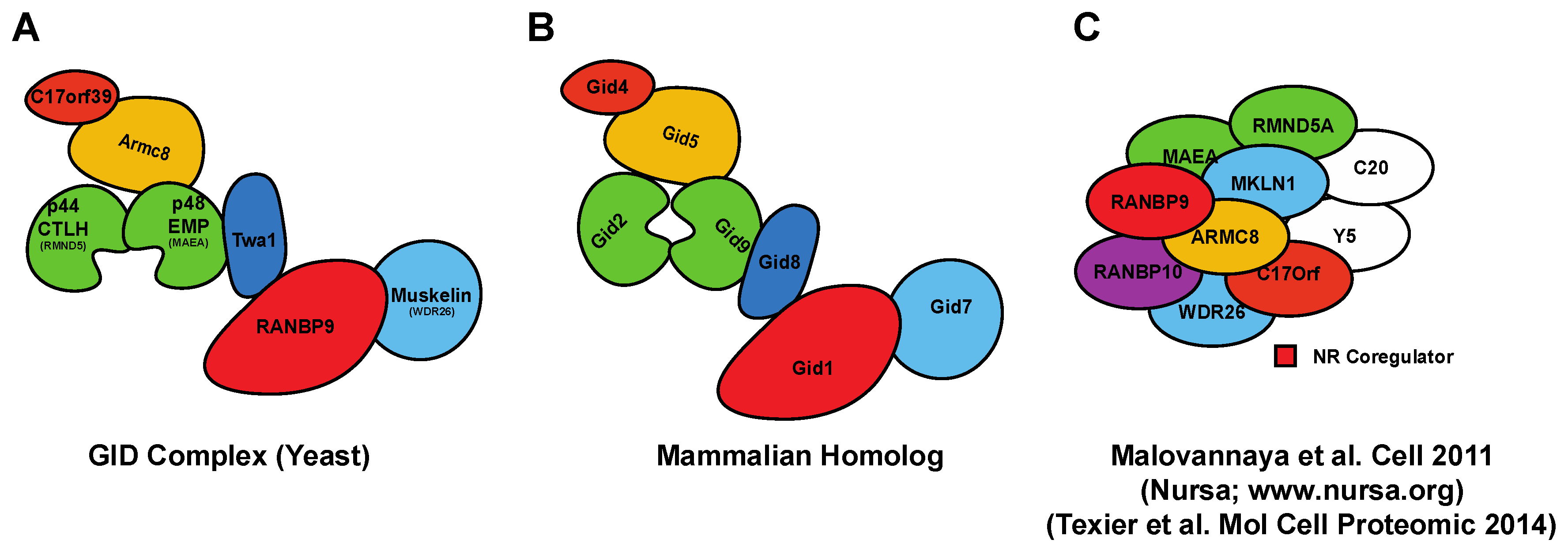
Figure 2.
Schematic representation of the S. cerevisiae Glucose-Induced degradation Deficient (GID)- and correspondent mammalian CTLH-macromolecular complexes. (A) The topology of the GID complex in yeast is well established [31]; (B) Predicted composition of the mammalian CTLH complex based on the GID mammalian homologs. The name of the complex comes from the CTLH domain that most of the members have; (C) CTLH or Nuclear Receptor coregulator-complex pulled down from mammalian cells including RANBP9 and RANBP10 (adapted from [29]). In the depicted complex, GID8 is named C20orf11 and indicated as C20. The experiment showed as part of the complex also YPEL5 (Yippee Like 5) indicated as Y5 in the cartoon, which has no known equivalent in the S. cerevisiae GID complex.
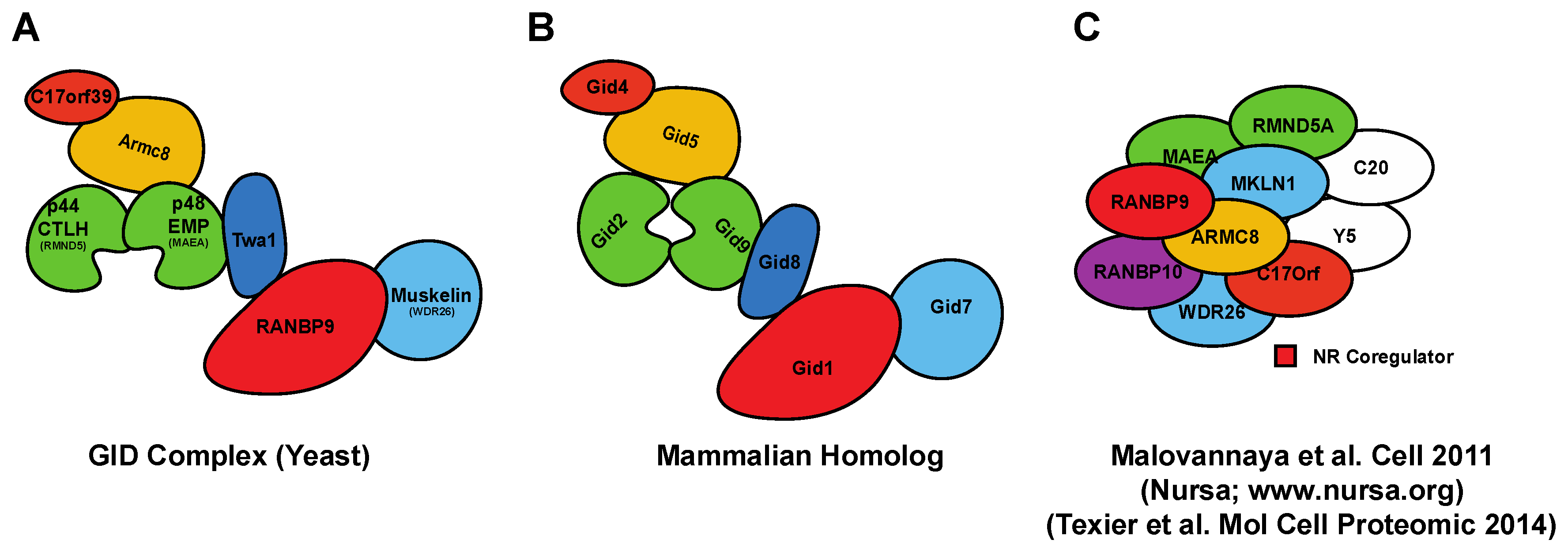
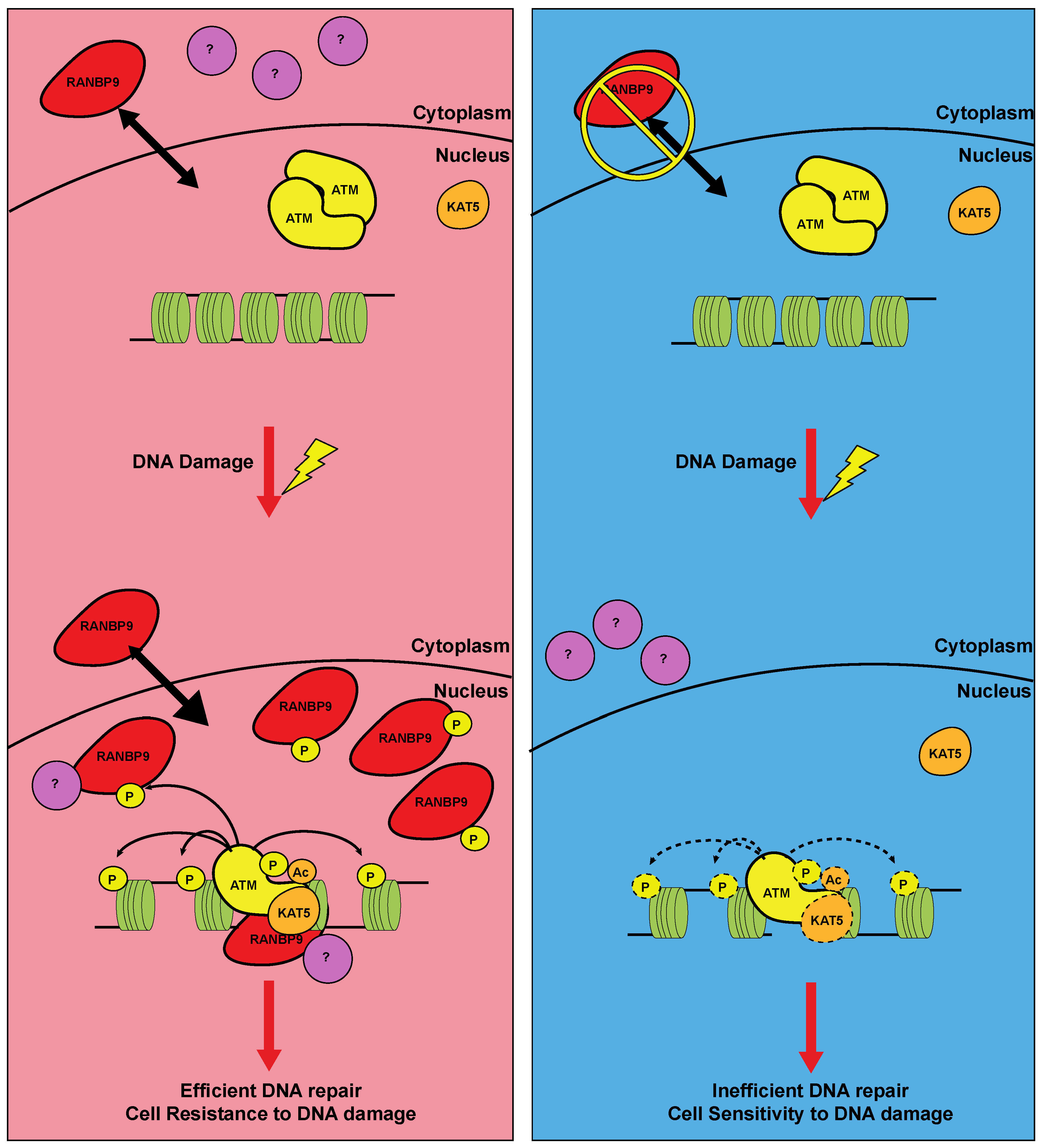
Figure 3.
Potential action mechanism of RANBP9 in ATM-dependent DDR. In RANBP9 (red) expressing cells and in the absence of DNA damage (left panel, top), RANBP9 protein shuttles between the nucleus and the cytoplasm. Upon DNA damage such as IR and DNA-damaging drugs (left panel, bottom), ATM is activated and enhances RANBP9 nuclear accumulation through its phosphorylation, potentially with other cytoplasmic partners (purple). This event potentially leads to enhanced KAT5-dependent ATM acetylation, a marker of its full activation. For this reason, RANBP9-expressing cells activate an efficient ATM signaling pathway, resulting in efficient DNA repair and survival to genotoxic stress. Conversely, when RANBP9 expression is reduced, the full activation of ATM is impaired, leading to inefficient DNA repair and sensitivity to DNA damaging agents.
Figure 3.
Potential action mechanism of RANBP9 in ATM-dependent DDR. In RANBP9 (red) expressing cells and in the absence of DNA damage (left panel, top), RANBP9 protein shuttles between the nucleus and the cytoplasm. Upon DNA damage such as IR and DNA-damaging drugs (left panel, bottom), ATM is activated and enhances RANBP9 nuclear accumulation through its phosphorylation, potentially with other cytoplasmic partners (purple). This event potentially leads to enhanced KAT5-dependent ATM acetylation, a marker of its full activation. For this reason, RANBP9-expressing cells activate an efficient ATM signaling pathway, resulting in efficient DNA repair and survival to genotoxic stress. Conversely, when RANBP9 expression is reduced, the full activation of ATM is impaired, leading to inefficient DNA repair and sensitivity to DNA damaging agents.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Phosphorylation sites by ATM, ATR and DNA-PK (PIK kinases) on RANBP9 and RANBP10. Selected list of serines and threonines (Amino acids “AA” column) and target peptides predicted by the algorithm Group-Based Prediction System, GPS 3.0 (http://gps.biocuckoo.org) to be phosphorylated by PIK-kinases on RANBP9 (Protein ID: Uniprot Q96S59) and RANBP10 (Protein ID: Uniprot Q6VN20). Protein domains in which the listed target sites are located are also indicated. Selection of predicted sites was based on a score > 2 and a difference between the score and the threshold >20% of the total score. The only exceptions to these criteria are RANBP9 S483 and RANBP10 S365, whose existence is reported in 58 and 160 datasets, respectively from the “PhosphoSitePlus” website. PhosphoSitePlus column lists the number of datasets in which the specific phosphorylation has been reported. For the references associated with those datasets and criteria used for the selection, please visit: www.phosphositeplus.org.
Table 1.
Phosphorylation sites by ATM, ATR and DNA-PK (PIK kinases) on RANBP9 and RANBP10. Selected list of serines and threonines (Amino acids “AA” column) and target peptides predicted by the algorithm Group-Based Prediction System, GPS 3.0 (http://gps.biocuckoo.org) to be phosphorylated by PIK-kinases on RANBP9 (Protein ID: Uniprot Q96S59) and RANBP10 (Protein ID: Uniprot Q6VN20). Protein domains in which the listed target sites are located are also indicated. Selection of predicted sites was based on a score > 2 and a difference between the score and the threshold >20% of the total score. The only exceptions to these criteria are RANBP9 S483 and RANBP10 S365, whose existence is reported in 58 and 160 datasets, respectively from the “PhosphoSitePlus” website. PhosphoSitePlus column lists the number of datasets in which the specific phosphorylation has been reported. For the references associated with those datasets and criteria used for the selection, please visit: www.phosphositeplus.org.
| Protein Domain | Residue | AA | Target Peptide | PhosphoSitePlus | |
|---|---|---|---|---|---|
| RANBP9 (Uniprot Q96S59) | PRY | 181 | S | KFSYIGLSQNNLRVH | 1 |
| LisH | 375 | S | MIQKMVSSYLVHHGY | - | |
| CTLH | 426 | T | MGEAIETTQQLYPSL | - | |
| Post-CTLH Region | 470 | S | LGGRSPKSQDSYPVS | 2 | |
| 483 | S | VSPRPFSSPSMSPSH | 58 | ||
| 550 | S | NSINMSRSQQVNNFT | 1 | ||
| 585 | S | NGFLNGSSKHDHEME | - | ||
| 603 | S | TEMEVDSSQLRRQLC | 2 | ||
| 613 | S | RRQLCGGSQAAIERM | 2 | ||
| CRA | 631 | S | GRELQAMSEQLRRDC | - | |
| 705 | T | ALAMGQATQCLGLMA | - | ||
| RANBP10 (Uniprot Q6VN20) | PRY | 69 | S | KYNYIGLSQGNLRVH | 1 |
| LisH | 263 | S | VLQNMVSSYLVHHGY | - | |
| CTLH | 314 | T | VGEAIETTQRFYPGL | - | |
| Post-CTLH Region | 358 | S | LSSRSPKSQDSYPGS | 5 | |
| 365 | S | SQDSYPGSPSLSPRH | 160 | ||
| 377 | S | PRHGPSSSHMHNTGA | - | ||
| 386 | S | MHNTGADSPSCSNGV | 7 | ||
| 439 | S | NSTDSTKSQHHSSTS | - | ||
| 490 | S | DLQTDESSMDDRHPR | 13 | ||
| CRA | 579 | S | LNSAILESQNLPKQP | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Palmieri, D.; Tessari, A.; Coppola, V. Scorpins in the DNA Damage Response. Int. J. Mol. Sci. 2018, 19, 1794. https://doi.org/10.3390/ijms19061794
AMA Style
Palmieri D, Tessari A, Coppola V. Scorpins in the DNA Damage Response. International Journal of Molecular Sciences. 2018; 19(6):1794. https://doi.org/10.3390/ijms19061794
Chicago/Turabian StylePalmieri, Dario, Anna Tessari, and Vincenzo Coppola. 2018. "Scorpins in the DNA Damage Response" International Journal of Molecular Sciences 19, no. 6: 1794. https://doi.org/10.3390/ijms19061794
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.