MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease

Abstract
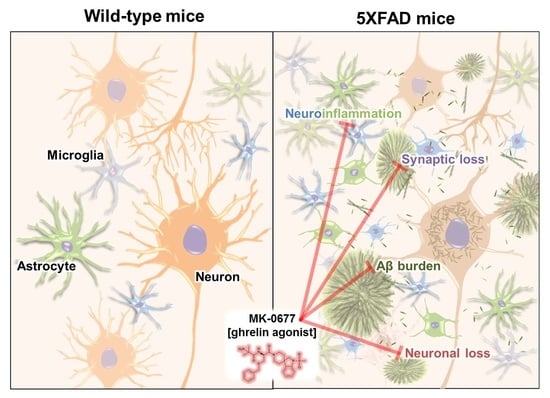
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Ghrelin Agonist Treatment Affected the Food Intake and Body Weight of the Healthy Animals
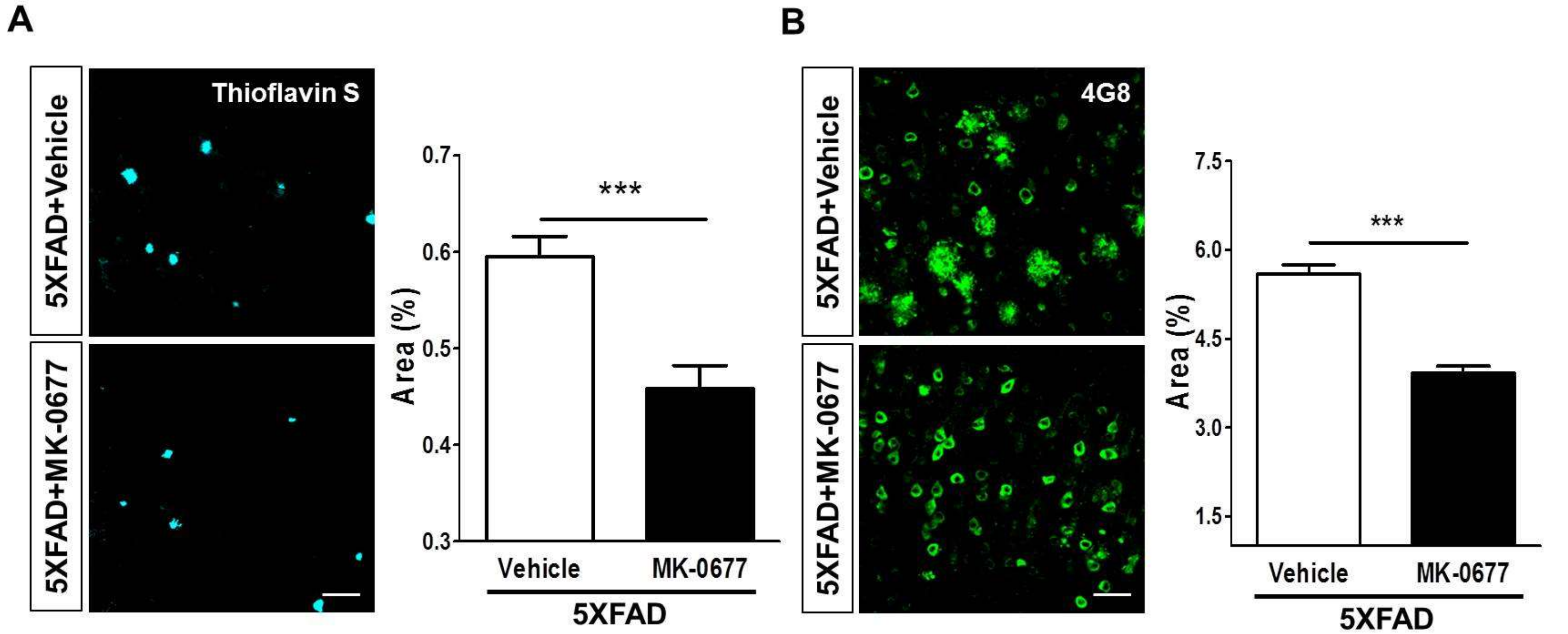
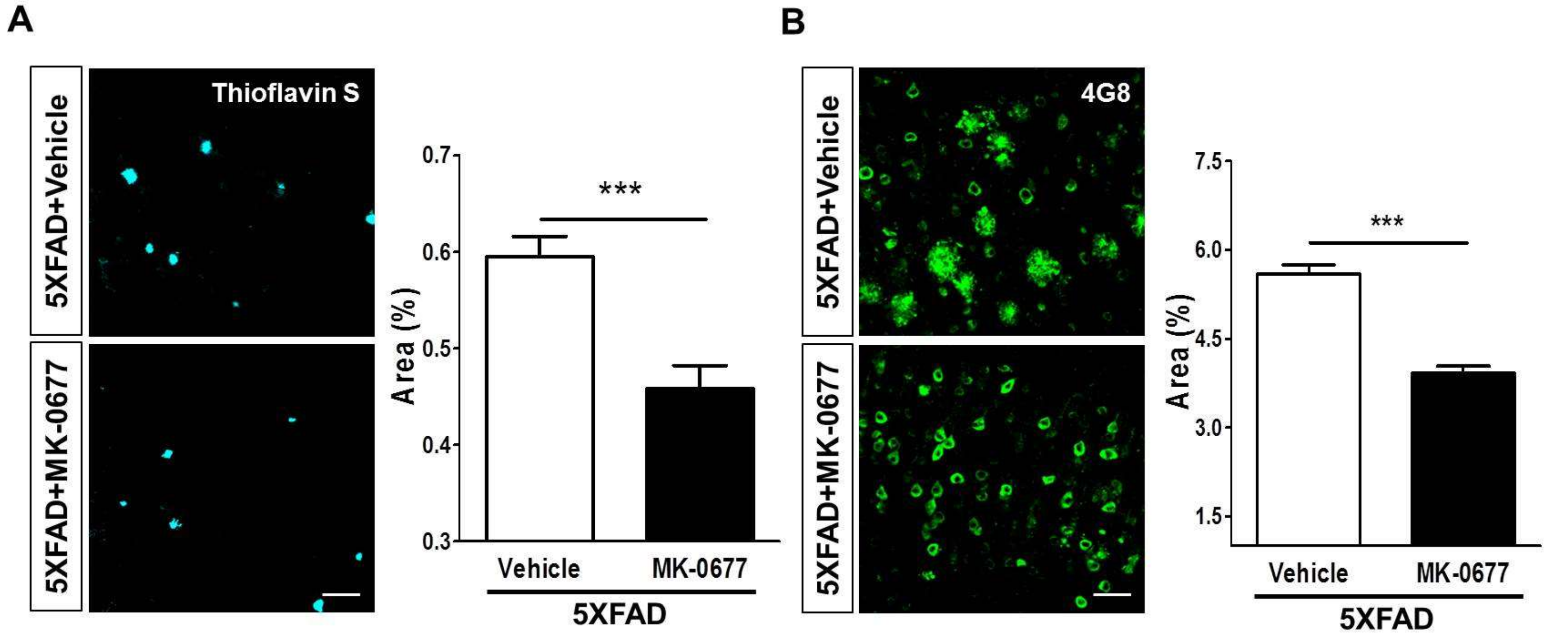
2.2. Ghrelin Agonist Treatment Significantly Reduces Aβ Accumulation in the Brains of 5XFAD Mice
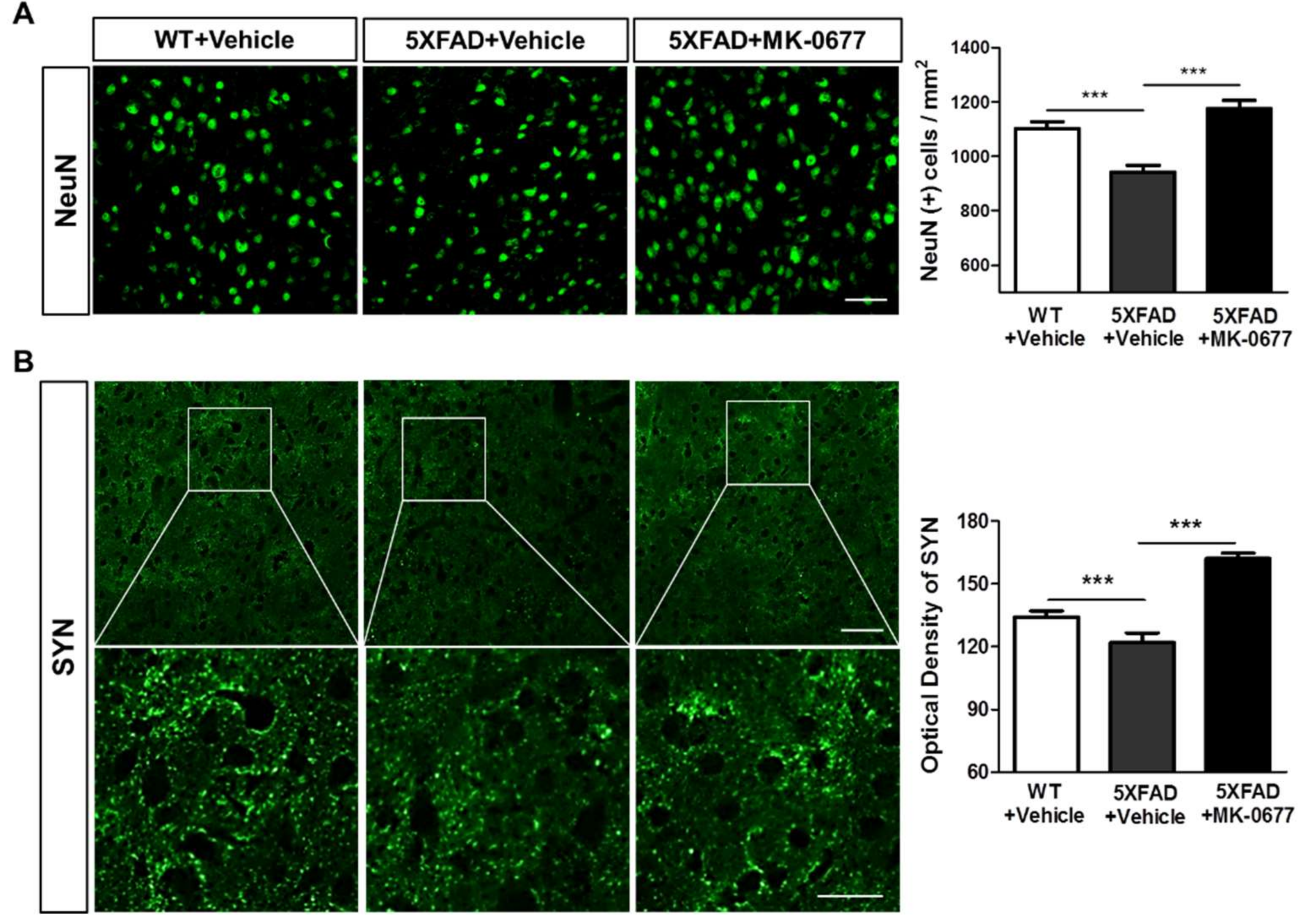
2.3. The Ghrelin Agonist Significantly Attenuated Neurodegeneration in the Neocortex of 5XFAD Mice
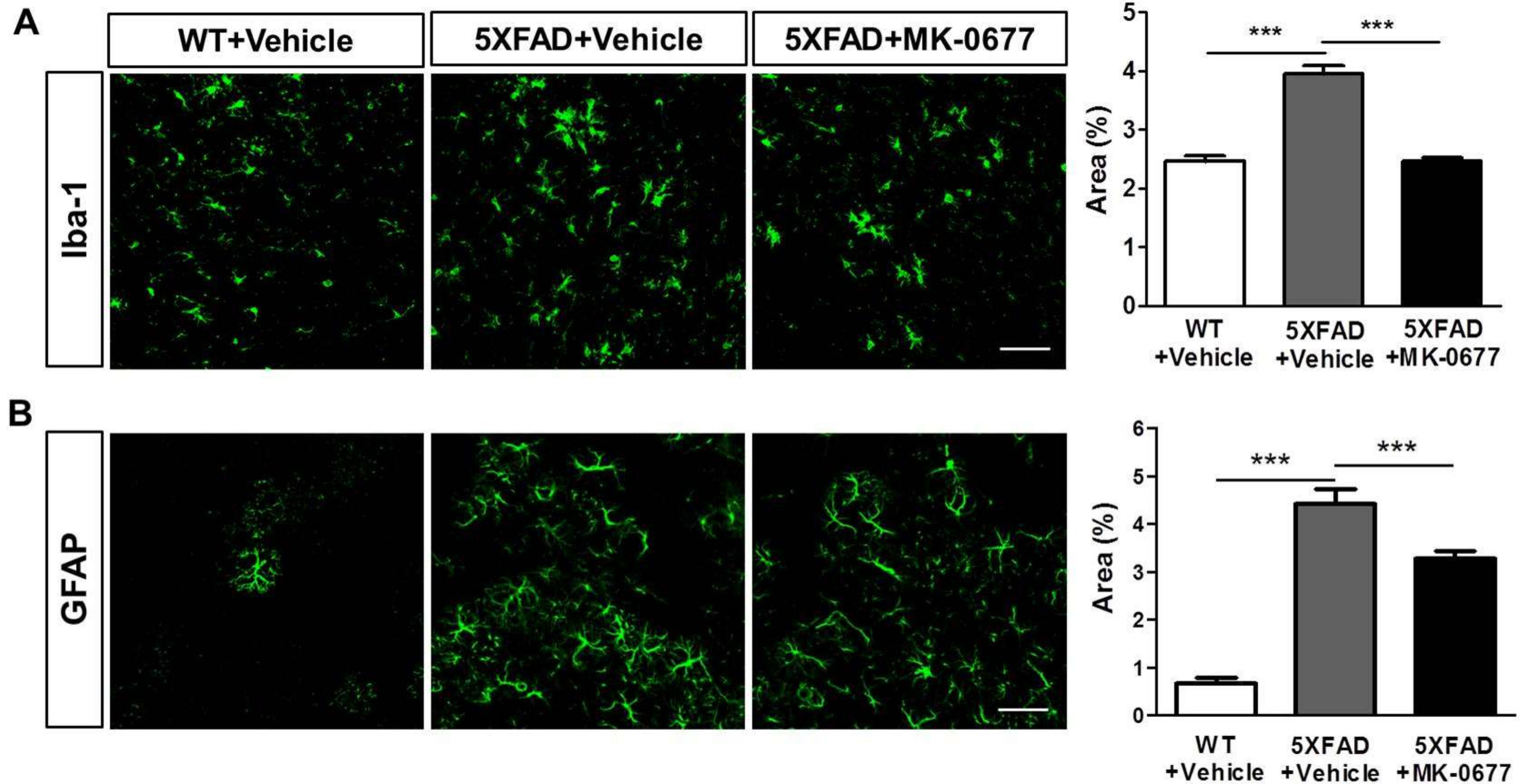
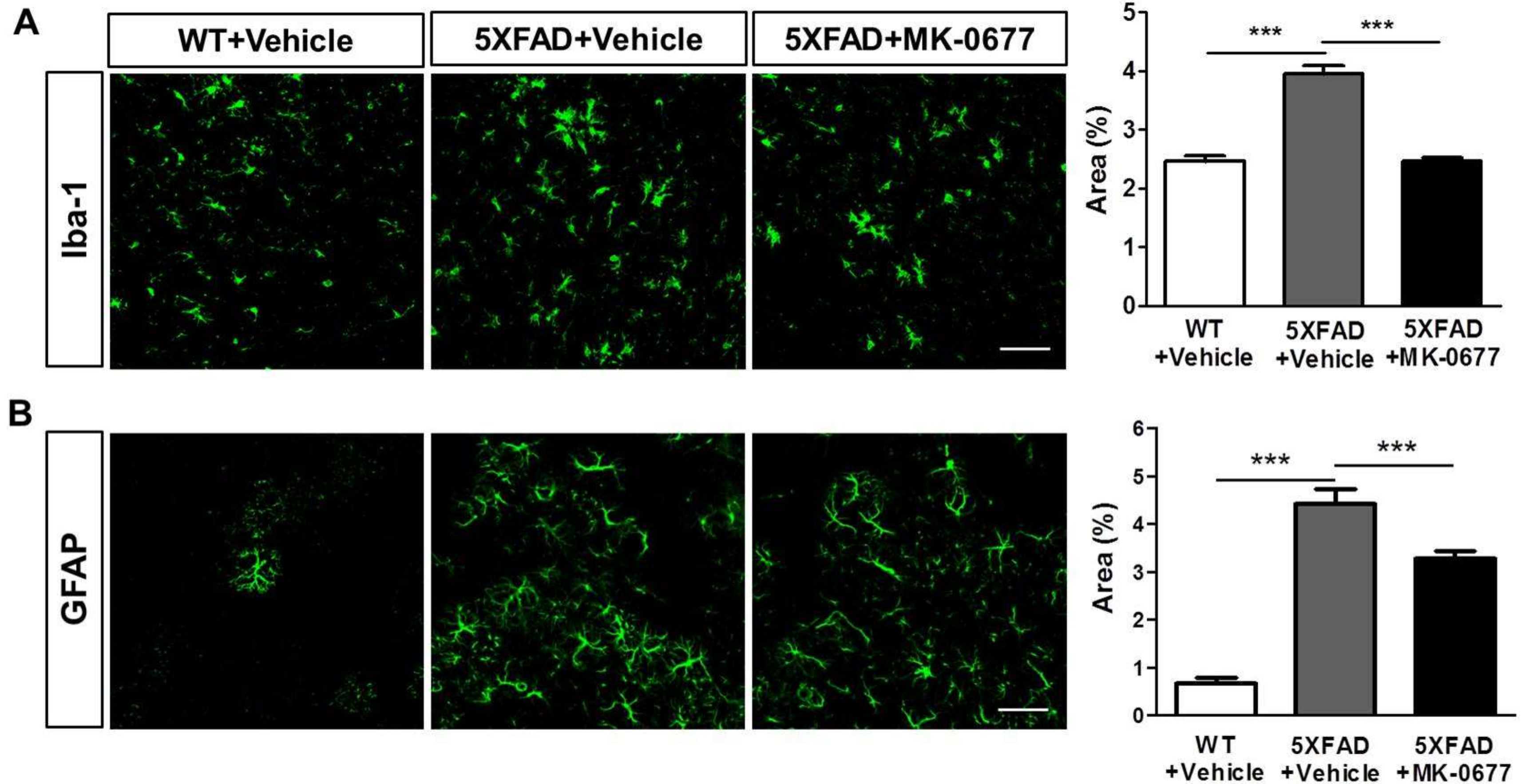
2.4. Ghrelin Agonist Significantly Inhibited Neuroinflammation in the Deep Cortical Layers of 5XFAD Mice
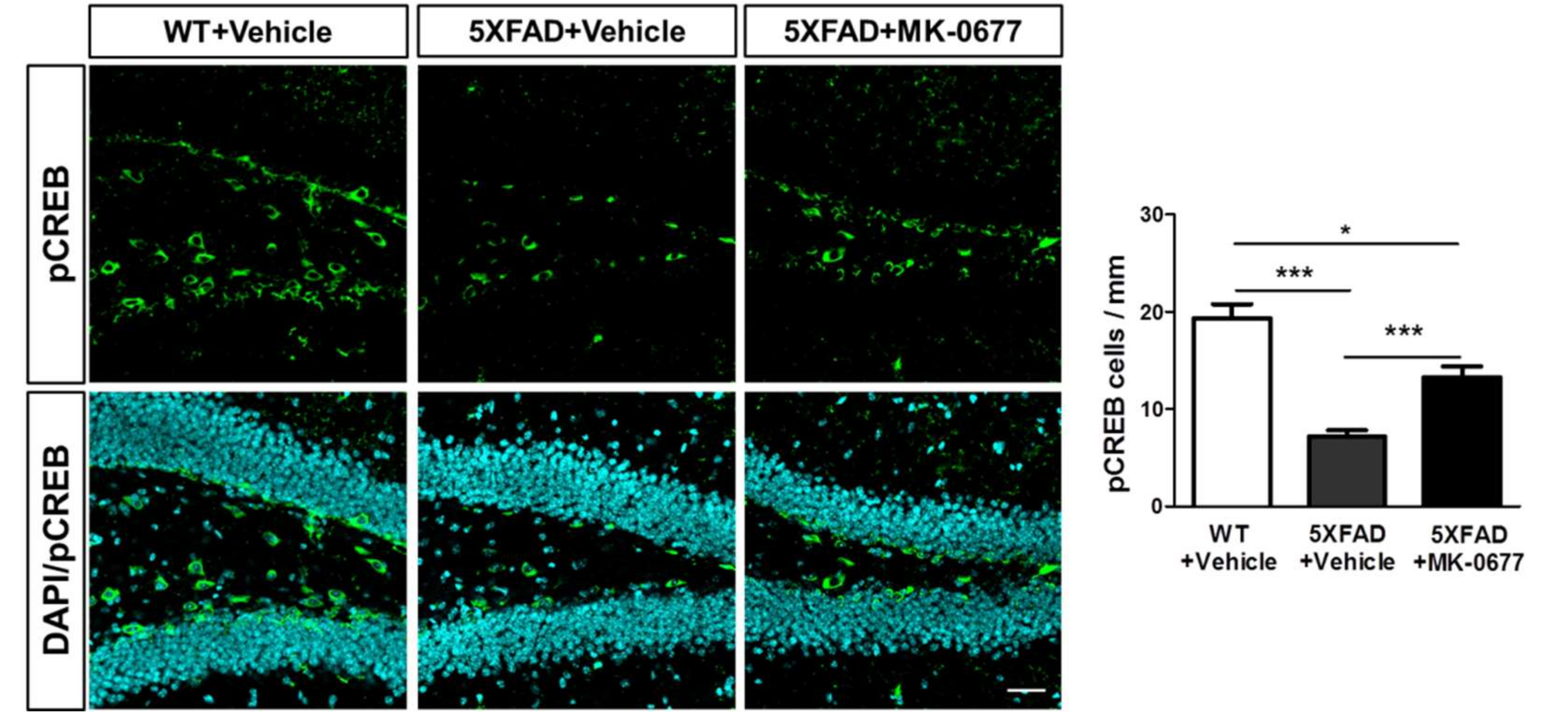
2.5. Ghrelin Agonist Significantly Increased Phosphorylation of CREB in Dentate Gyrus of the Hippocampus of 5XFAD Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Drug Treatment
4.2. Brain Tissue Preparation
4.3. Thioflavin-S Staining
4.4. Immunofluorescence Labeling
4.5. Image Acquisition and Quantification
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar]
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimers Dement. 2017, 13, 325–373. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Ariyasu, H.; Takaya, K.; Tagami, T.; Ogawa, Y.; Hosoda, K.; Akamizu, T.; Suda, M.; Koh, T.; Natsui, K.; Toyooka, S.; et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J. Clin. Endocrinol. Metab. 2001, 86, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Delhanty, P.J.; Neggers, S.J.; van der Lely, A.J. Mechanisms in endocrinology: Ghrelin: The differences between acyl- and des-acyl ghrelin. Eur. J. Endocrinol. 2012, 167, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Hayashida, Y.; Nakao, N.; Nakai, N.; Nakashima, K. Testis-specific and developmentally induced expression of a ghrelin gene-derived transcript that encodes a novel polypeptide in the mouse. Biochim. Biophys. Acta 2001, 1522, 62–65. [Google Scholar] [CrossRef]
- Caminos, J.E.; Tena-Sempere, M.; Gaytan, F.; Sanchez-Criado, J.E.; Barreiro, M.L.; Nogueiras, R.; Casanueva, F.F.; Aguilar, E.; Dieguez, C. Expression of ghrelin in the cyclic and pregnant rat ovary. Endocrinology 2003, 144, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Yoshimoto, A.; Takaya, K.; Hosoda, K.; Ariyasu, H.; Yahata, K.; Mukoyama, M.; Sugawara, A.; Hosoda, H.; Kojima, M.; et al. Kidney produces a novel acylated peptide, ghrelin. FEBS Lett. 2000, 486, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Korbonits, M.; Bustin, S.A.; Kojima, M.; Jordan, S.; Adams, E.F.; Lowe, D.G.; Kangawa, K.; Grossman, A.B. The expression of the growth hormone secretagogue receptor ligand ghrelin in normal and abnormal human pituitary and other neuroendocrine tumors. J. Clin. Endocrinol. Metab. 2001, 86, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Saito, T.; Yagyu, T.; Jiang, B.H.; Kitagawa, K.; Inagaki, C. GH, GH receptor, GH secretagogue receptor, and ghrelin expression in human T cells, B cells, and neutrophils. J. Clin. Endocrinol. Metab. 2001, 86, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Volante, M.; Allia, E.; Gugliotta, P.; Funaro, A.; Broglio, F.; Deghenghi, R.; Muccioli, G.; Ghigo, E.; Papotti, M. Expression of ghrelin and of the GH secretagogue receptor by pancreatic islet cells and related endocrine tumors. J. Clin. Endocrinol. Metab. 2002, 87, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Guan, J.L.; Wang, Q.P.; Uehara, K.; Yamada, S.; Goto, N.; Date, Y.; Nakazato, M.; Kojima, M.; Kangawa, K.; et al. Immunocytochemical observation of ghrelin-containing neurons in the rat arcuate nucleus. Neurosci. Lett. 2002, 321, 157–160. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschop, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Hou, Z.; Miao, Y.; Gao, L.; Pan, H.; Zhu, S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul. Pept. 2006, 134, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Cordoba-Chacon, J.; Kineman, R.D.; Luque, R.M.; Castano, J.P. Role of ghrelin system in neuroprotection and cognitive functions: Implications in Alzheimer’s disease. Peptides 2011, 32, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, S.; Park, S. Neurogenic Effects of Ghrelin on the Hippocampus. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Rubio, A.; Cordoba-Chacon, J.; Gracia-Navarro, F.; Kineman, R.D.; Avila, J.; Luque, R.M.; Castano, J.P. Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 2010, 22, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, A.; Metallinos, I.C.; Psyrogiannis, A.; Vagenakis, G.A.; Kyriazopoulou, V. Ghrelin and leptin secretion in patients with moderate Alzheimer’s disease. J. Nutr. Health Aging 2012, 16, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, G.; Samson, S.L.; Sun, Y. Ghrelin: Much more than a hunger hormone. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Heppner, K.M.; Piechowski, C.L.; Muller, A.; Ottaway, N.; Sisley, S.; Smiley, D.L.; Habegger, K.M.; Pfluger, P.T.; Dimarchi, R.; Biebermann, H.; et al. Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes 2014, 63, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Gil-Campos, M.; Aguilera, C.M.; Canete, R.; Gil, A. Ghrelin: A hormone regulating food intake and energy homeostasis. Br. J. Nutr. 2006, 96, 201–226. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ukkola, O.; Poykko, S. Ghrelin, growth and obesity. Ann. Med. 2002, 34, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, I.I.; Hofmeijer, J.; van Putten, M.; le Feber, J. Acyl Ghrelin Improves Synapse Recovery in an In Vitro Model of Postanoxic Encephalopathy. Mol. Neurobiol. 2016, 53, 6136–6143. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kim, E.; Lee, D.H.; Seo, S.; Ju, S.; Lee, D.; Kim, H.; Park, S. Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 2007, 148, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Li, E.; Kim, Y.; Kim, S.; Park, S. Multiple signaling pathways mediate ghrelin-induced proliferation of hippocampal neural stem cells. J. Endocrinol. 2013, 218, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Hu, S.; Wu, B.; Miao, Y.; Pan, H.; Zhu, S. Ghrelin inhibits apoptosis signal-regulating kinase 1 activity via upregulating heat-shock protein 70. Biochem. Biophys. Res. Commun. 2007, 359, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, I.; Tamiazzo, L.; Bresciani, E.; Rapetti, D.; Caporali, S.; Lattuada, D.; Locatelli, V.; Torsello, A. Desacyl-ghrelin and synthetic GH-secretagogues modulate the production of inflammatory cytokines in mouse microglia cells stimulated by beta-amyloid fibrils. J. Neurosci. Res. 2009, 87, 2718–2727. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, P.S.; Xie, D.; Liu, K.; Chen, L. Ghrelin reduces injury of hippocampal neurons in a rat model of cerebral ischemia/reperfusion. Chin. J. Physiol. 2006, 49, 244–250. [Google Scholar] [PubMed]
- Xu, J.; Wang, S.; Lin, Y.; Cao, L.; Wang, R.; Chi, Z. Ghrelin protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci. Lett. 2009, 453, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschoep, M.; et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A.; et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Chung, H.; Kim, Y.; Kim, D.H.; Ryu, J.H.; Sato, T.; Kojima, M.; Park, S. Ghrelin directly stimulates adult hippocampal neurogenesis: Implications for learning and memory. Endocr. J. 2013, 60, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Choi, J.G.; Nam, D.W.; Hong, H.S.; Choi, Y.J.; Oh, M.S.; Mook-Jung, I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1-42 oligomer-injected mice. J. Alzheimers Dis. 2011, 23, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimers Dis. 2014, 41, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.R.; Smith, R.G.; Leng, G. The nonpeptide growth hormone secretagogue, MK-0677, activates hypothalamic arcuate nucleus neurons in vivo. J. Neuroendocrinol. 1998, 10, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Patchett, A.A.; Nargund, R.P.; Tata, J.R.; Chen, M.H.; Barakat, K.J.; Johnston, D.B.; Cheng, K.; Chan, W.W.; Butler, B.; Hickey, G.; et al. Design and biological activities of L-163,191 (MK-0677): A potent, orally active growth hormone secretagogue. Proc. Natl. Acad. Sci. USA 1995, 92, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.A.; Langmead, C.J.; Wise, A.; Milligan, G. Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super-agonists but not allosteric regulators for activation of the G protein Galpha(o1) by the Ghrelin receptor. Mol. Pharmacol. 2009, 76(4), 802–811. [Google Scholar] [CrossRef] [PubMed]
- Howick, K.; Griffin, B.T.; Cryan, J.F.; Schellekens, H. From Belly to Brain: Targeting the Ghrelin Receptor in Appetite and Food Intake Regulation. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Tolle, V.; Bassant, M.H.; Zizzari, P.; Poindessous-Jazat, F.; Tomasetto, C.; Epelbaum, J.; Bluet-Pajot, M.T. Ultradian rhythmicity of ghrelin secretion in relation with GH, feeding behavior, and sleep-wake patterns in rats. Endocrinology 2002, 143, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Chapman, I.M.; Bach, M.A.; Van Cauter, E.; Farmer, M.; Krupa, D.; Taylor, A.M.; Schilling, L.M.; Cole, K.Y.; Skiles, E.H.; Pezzoli, S.S.; et al. Stimulation of the growth hormone (GH)-insulin-like growth factor I axis by daily oral administration of a GH secretogogue (MK-677) in healthy elderly subjects. J. Clin. Endocrinol. Metab. 1996, 81, 4249–4257. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, E.J.; Allison, D.B.; van Groen, T.; Kadish, I. Hunger in the absence of caloric restriction improves cognition and attenuates Alzheimer’s disease pathology in a mouse model. PLoS ONE 2013, 8, e60437. [Google Scholar] [CrossRef] [PubMed]
- Kunath, N.; van Groen, T.; Allison, D.B.; Kumar, A.; Dozier-Sharpe, M.; Kadish, I. Ghrelin agonist does not foster insulin resistance but improves cognition in an Alzheimer’s disease mouse model. Sci. Rep. 2015, 5, 11452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhavadas, S.; Kutty, B.M.; Subramanian, S. Amyloid beta lowering and cognition enhancing effects of ghrelin receptor analog [d-Lys (3)] GHRP-6 in rat model of obesity. Indian J. Biochem. Biophys. 2014, 51, 257–262. [Google Scholar] [PubMed]
- Sevigny, J.J.; Ryan, J.M.; van Dyck, C.H.; Peng, Y.; Lines, C.R.; Nessly, M.L.; Group, M.K.P.S. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology 2008, 71, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Bartolotti, N.; Segura, L.; Lazarov, O. Diminished CRE-Induced Plasticity is Linked to Memory Deficits in Familial Alzheimer’s Disease Mice. J. Alzheimers Dis. 2016, 50, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Martinez, S. A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 2015, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Joharapurkar, A.; Dhanesha, N.; Patel, V.; Kshirsagar, S.; Raval, P.; Raval, S.; Jain, M.R. Thyroid hormone modulates food intake and glycemia via ghrelin secretion in Zucker fatty rats. Drug Res. (Stuttg.) 2014, 64, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Geng, J.; Bao, J.; Tang, Y.; Liu, M.; Yu, H.; Han, Y.; Huang, W.; Zhou, S. Two ghrelin receptor agonists for adults with malnutrition: A systematic review and meta-analysis. Nutr. J. 2016, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Swerdloff, R.; Wang, C.; Kyle, M.; Kipnes, M.; Biller, B.M.; Cook, D.; Yuen, K.C.; Bonert, V.; Dobs, A.; et al. Macimorelin (AEZS-130)-stimulated growth hormone (GH) test: Validation of a novel oral stimulation test for the diagnosis of adult GH deficiency. J. Clin. Endocrinol. Metab. 2013, 98, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Charoenthongtrakul, S.; Giuliana, D.; Longo, K.A.; Govek, E.K.; Nolan, A.; Gagne, S.; Morgan, K.; Hixon, J.; Flynn, N.; Murphy, B.J.; et al. Enhanced gastrointestinal motility with orally active ghrelin receptor agonists. J. Pharmacol. Exp. Therap. 2009, 329, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.A.; Patrie, J.T.; Gaylinn, B.D.; Thorner, M.O.; Bolton, W.K. Oral ghrelin receptor agonist MK-0677 increases serum insulin-like growth factor 1 in hemodialysis patients: A randomized blinded study. Nephrol. Dialysis Transpl.: Off. Publ. Eur. Dialysis Transpl. Assoc.—Eur. Renal Assoc. 2018, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.D.; Baranger, K.; Gauthier, C.; Jacquet, M.; Bernard, A.; Escoffier, G.; Marchetti, E.; Khrestchatisky, M.; Rivera, S.; Roman, F.S. Evidence for early cognitive impairment related to frontal cortex in the 5XFAD mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Ohno, M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 2009, 33, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, M. Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol. Learn. Memory 2009, 92, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczorowski, C.C.; Sametsky, E.; Shah, S.; Vassar, R.; Disterhoft, J.F. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur. J. Neurosci. 2010, 31, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.D.; Jacquet, M.; Baranger, K.; Migliorati, M.; Escoffier, G.; Bernard, A.; Khrestchatisky, M.; Feron, F.; Rivera, S.; Roman, F.S.; et al. Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer’s disease. Hippocampus 2014, 24, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Isokawa, M. The role of ghrelin in the hippocampal neuron plasticity. Alzheimers Dement.: J. Alzheimers Assoc. 2009, 5, P172. [Google Scholar] [CrossRef]
- Yamamoto-Sasaki, M.; Ozawa, H.; Saito, T.; Rosler, M.; Riederer, P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999, 824, 300–303. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons. Mol. Neurodegen. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Bayer, T.A. Intraneuronal Aβ accumulation and neurodegeneration: Lessons from transgenic models. Life Sci. 2012, 91, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Kim, H.G.; Hwang, L.; Seo, J.H.; Kim, S.; Hwang, S.; Kim, S.; Lee, D.; Chung, H.; Oh, M.S.; et al. Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease by blocking microglial activation. Neurotoxicity Res. 2009, 15, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yune, T.Y. Ghrelin inhibits oligodendrocyte cell death by attenuating microglial activation. Endocrinol. Metab. (Seoul Korea) 2014, 29, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Fuente-Martin, E.; Diaz, F.; Granado, M.; Argente-Arizon, P.; Frago, L.M.; Freire-Regatillo, A.; Barrios, V.; Argente, J.; Chowen, J.A. The opposing effects of ghrelin on hypothalamic and systemic inflammatory processes are modulated by its acylation status and food intake in male rats. Endocrinology 2014, 155, 2868–2880. [Google Scholar] [CrossRef] [PubMed]
- Inui, A. Ghrelin: An orexigenic and somatotrophic signal from the stomach. Nat. Rev. Neurosci. 2001, 2, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Trumbauer, M.E.; Chen, A.S.; Weingarth, D.T.; Adams, J.R.; Frazier, E.G.; Shen, Z.; Marsh, D.J.; Feighner, S.D.; Guan, X.M.; et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 2004, 145, 2607–2612. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.G.; Plunkett, L.M.; Gertz, B.J.; He, W.; Wittreich, J.; Polvino, W.M.; Clemmons, D.R. MK-677, an orally active growth hormone secretagogue, reverses diet-induced catabolism. J. Clin. Endocrinol. Metab. 1998, 83, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Nass, R.; Pezzoli, S.S.; Oliveri, M.C.; Patrie, J.T.; Harrell, F.E., Jr.; Clasey, J.L.; Heymsfield, S.B.; Bach, M.A.; Vance, M.L.; Thorner, M.O. Effects of an oral ghrelin mimetic on body composition and clinical outcomes in healthy older adults: A randomized trial. Ann. Internal Med. 2008, 149, 601–611. [Google Scholar] [CrossRef]
- Caberlotto, L.; Lauria, M.; Nguyen, T.P.; Scotti, M. The central role of AMP-kinase and energy homeostasis impairment in Alzheimer’s disease: A multifactor network analysis. PLoS ONE 2013, 8, e78919. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of β-amyloid induced Tau-pathology: The long and “folded” road to understand the mechanism. Mol. Neurodegener. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Tsuchiya, A.; Nishizaki, T. Hyperphosphorylation of Tau at Ser396 occurs in the much earlier stage than appearance of learning and memory disorders in 5XFAD mice. Behav. Brain Res. 2014, 274, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, C.L.; Kokjohn, T.A.; Whiteside, C.M.; Macias, M.P.; Kalback, W.M.; Sabbagh, M.N.; Beach, T.G.; Vassar, R.; Roher, A.E. Molecular Differences and Similarities Between Alzheimer’s Disease and the 5XFAD Transgenic Mouse Model of Amyloidosis. Biochem. Insights 2013, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hastie, C.J.; McLauchlan, H.; Cohen, P.; Goedert, M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 2004, 90(2), 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.; Lee, S.; Li, E.; Kim, Y.; Park, S. Ghrelin protects spinal cord motoneurons against chronic glutamate-induced excitotoxicity via ERK1/2 and phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3beta pathways. Exp. Neurol. 2011, 230, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zeng, M.; He, W.; Huang, X.; Luo, L.; Zhang, H.; Deng, D.Y. Ghrelin protects alveolar macrophages against lipopolysaccharide-induced apoptosis through growth hormone secretagogue receptor 1a-dependent c-Jun N-terminal kinase and Wnt/beta-catenin signaling and suppresses lung inflammation. Endocrinology 2015, 156, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Huang, J.; Li, H.; Yang, Z.; Zeng, Y.; Liu, J.; Hao, Y.; Li, R. Ghrelin accelerates wound healing through GHS-R1a-mediated MAPK-NF-kappaB/GR signaling pathways in combined radiation and burn injury in rats. Sci. Rep. 2016, 6, 27499. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wang, J.; Yu, F.; Li, Z.; Li, H.; Guo, C.; Fan, X. Ghrelin protects against palmitic acid or lipopolysaccharide-induced hepatocyte apoptosis through inhibition of MAPKs/iNOS and restoration of Akt/eNOS pathways. Biomed. Pharmacother. 2016, 84, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, G.; Wang, Q.; Guo, F.; Wang, H. Acylated ghrelin protects hippocampal neurons in pilocarpine-induced seizures of immature rats by inhibiting cell apoptosis. Mol. Biol. Rep. 2013, 40, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Park, S. Ghrelin regulates cell cycle-related gene expression in cultured hippocampal neural stem cells. J. Endocrinol. 2016, 230, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Kim, Y.; Kim, S.; Sato, T.; Kojima, M.; Park, S. Ghrelin stimulates proliferation, migration and differentiation of neural progenitors from the subventricular zone in the adult mice. Exp. Neurol. 2014, 252, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Paxinos, G. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Elsevier/Academic Press: Boston, MA, USA, 2013. [Google Scholar]
- Jeon, S.G.; Kim, Y.J.; Kim, K.A.; Mook-Jung, I.; Moon, M. Visualization of Altered Hippocampal Connectivity in an Animal Model of Alzheimer’s Disease. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.-o.; Shin, S.J.; Park, J.Y.; Ku, B.K.; Song, J.S.; Kim, J.-J.; Jeon, S.G.; Lee, S.M.; Moon, M. MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1800. https://doi.org/10.3390/ijms19061800
Jeong Y-o, Shin SJ, Park JY, Ku BK, Song JS, Kim J-J, Jeon SG, Lee SM, Moon M. MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(6):1800. https://doi.org/10.3390/ijms19061800
Chicago/Turabian StyleJeong, Yu-on, Soo Jung Shin, Jun Yong Park, Bo Kyeong Ku, Ji Soo Song, Jwa-Jin Kim, Seong Gak Jeon, Sang Min Lee, and Minho Moon. 2018. "MK-0677, a Ghrelin Agonist, Alleviates Amyloid Beta-Related Pathology in 5XFAD Mice, an Animal Model of Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 6: 1800. https://doi.org/10.3390/ijms19061800