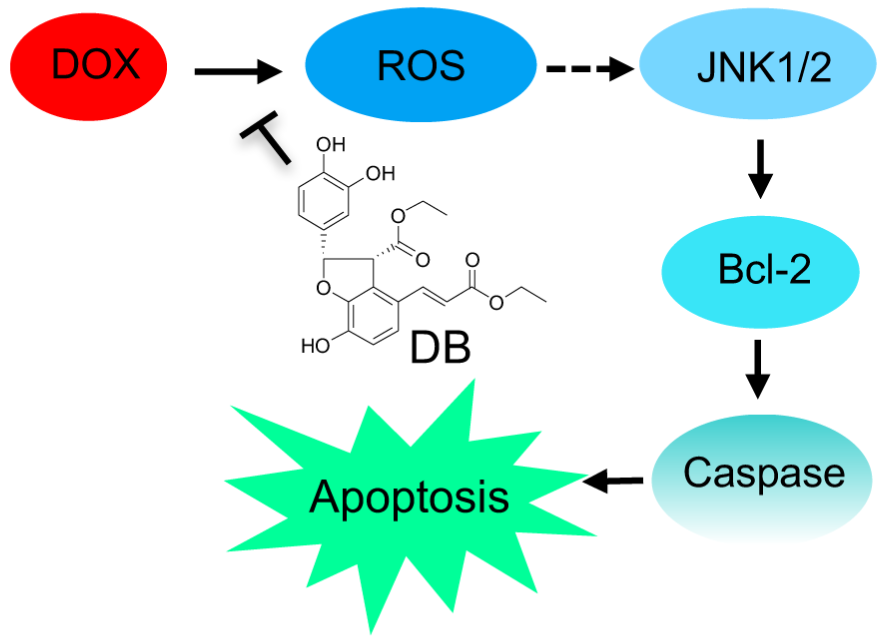
Diethyl Blechnic, a Novel Natural Product Isolated from Salvia miltiorrhiza Bunge, Inhibits Doxorubicin-Induced Apoptosis by Inhibiting ROS and Activating JNK1/2


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
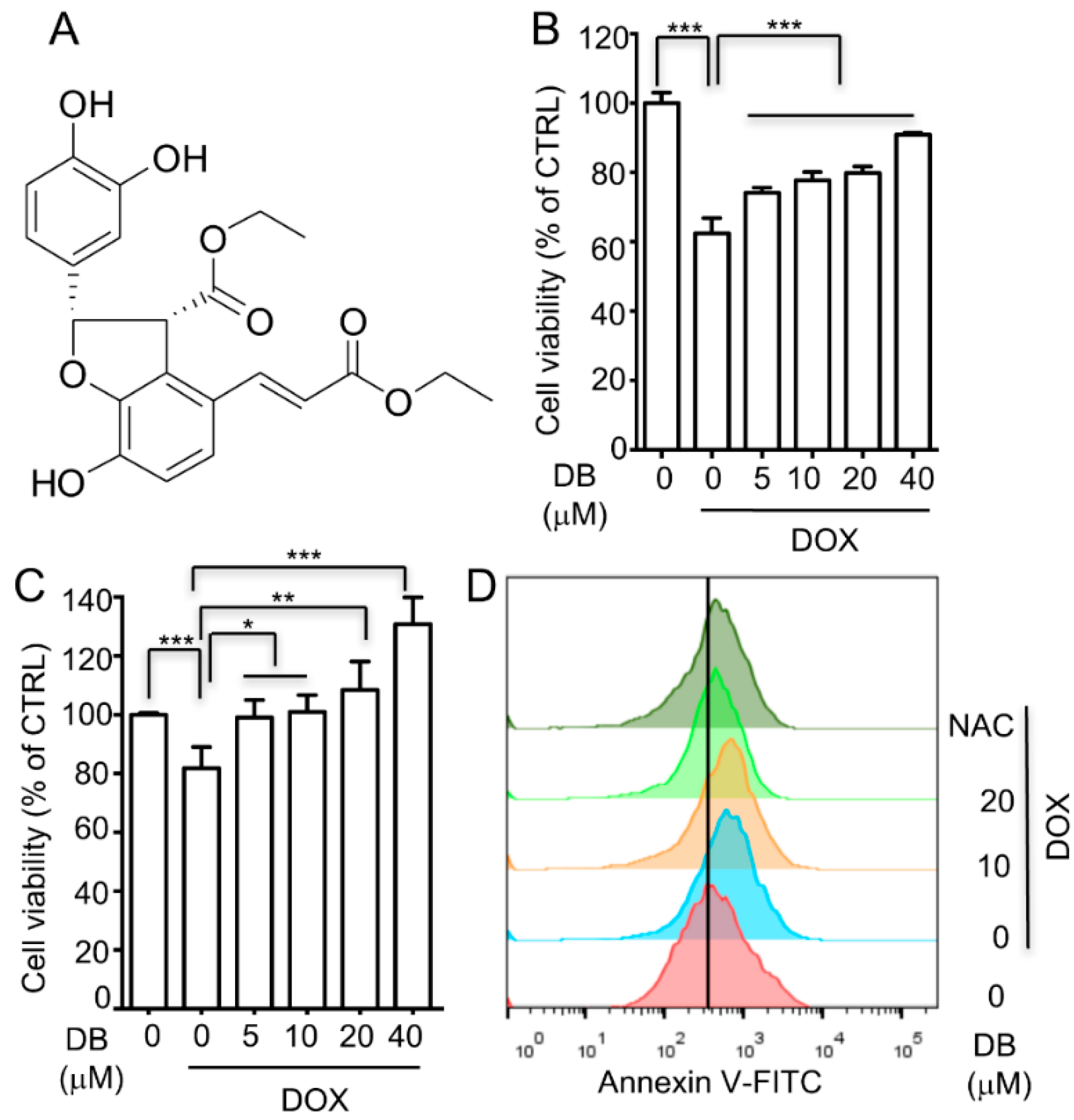
2.1. DB Protects Cells from DOX-Induced Cell Death
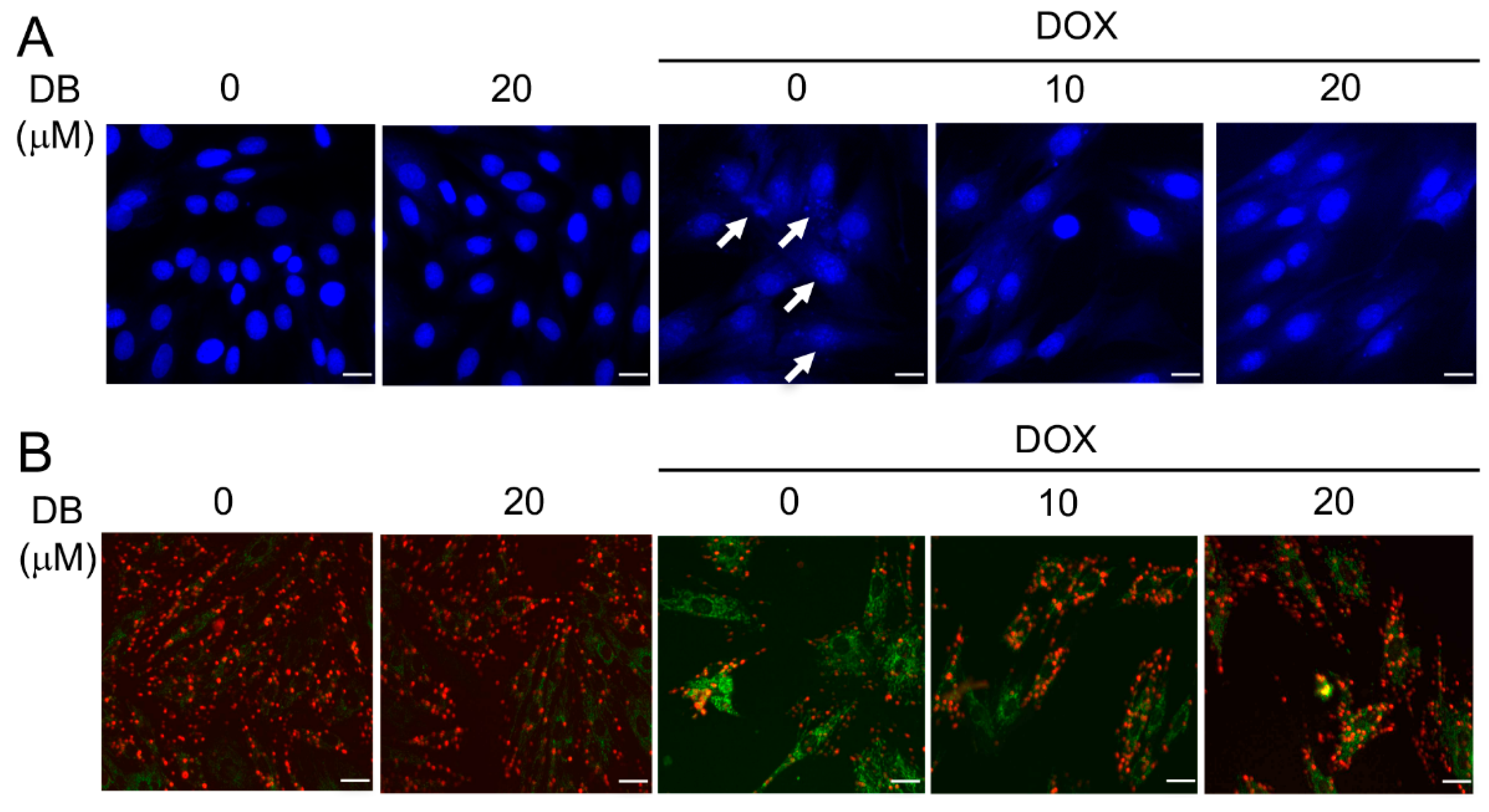
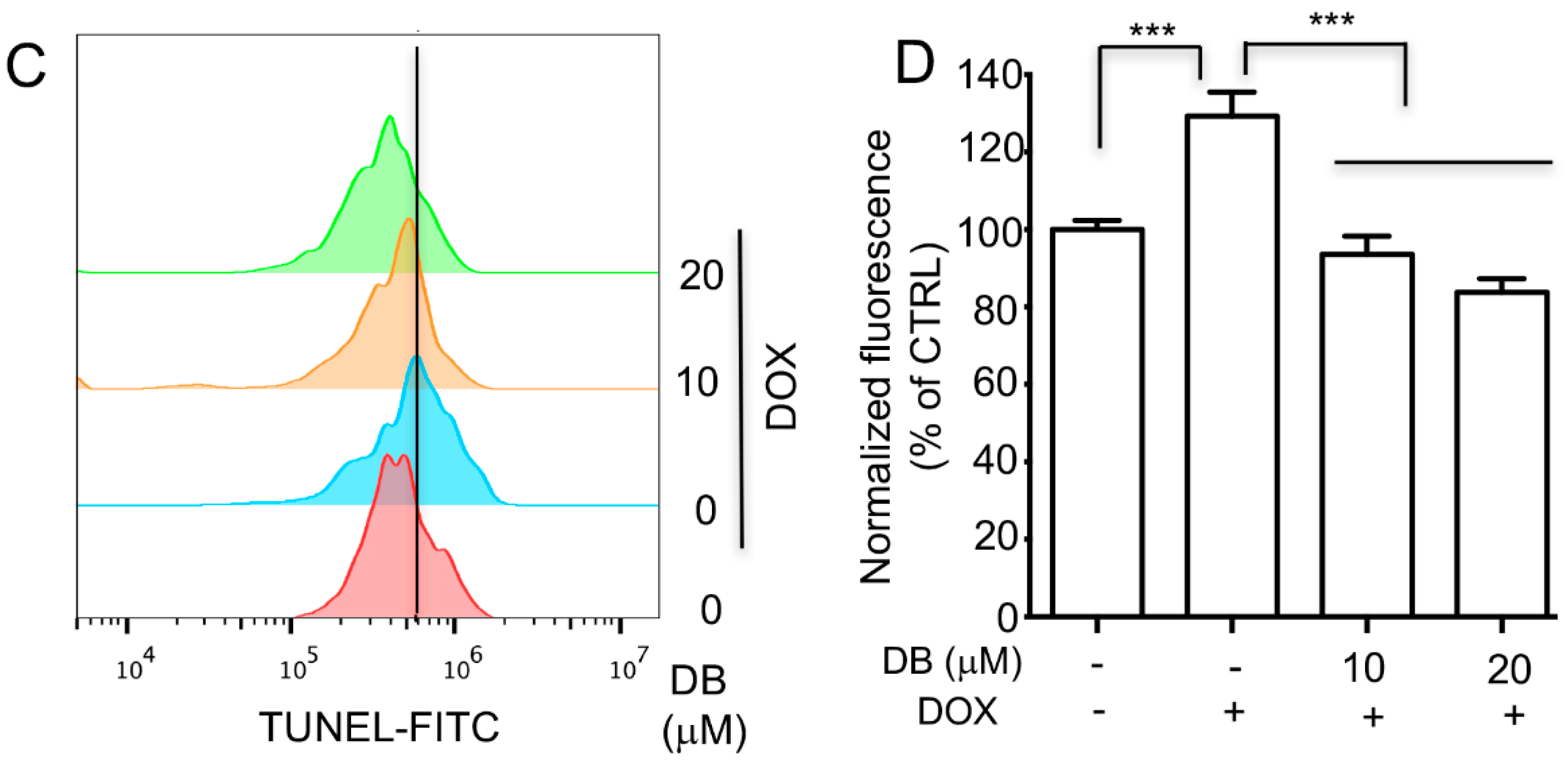
2.2. DB Protects from DOX-Induced Cardiac Cell Apoptosis
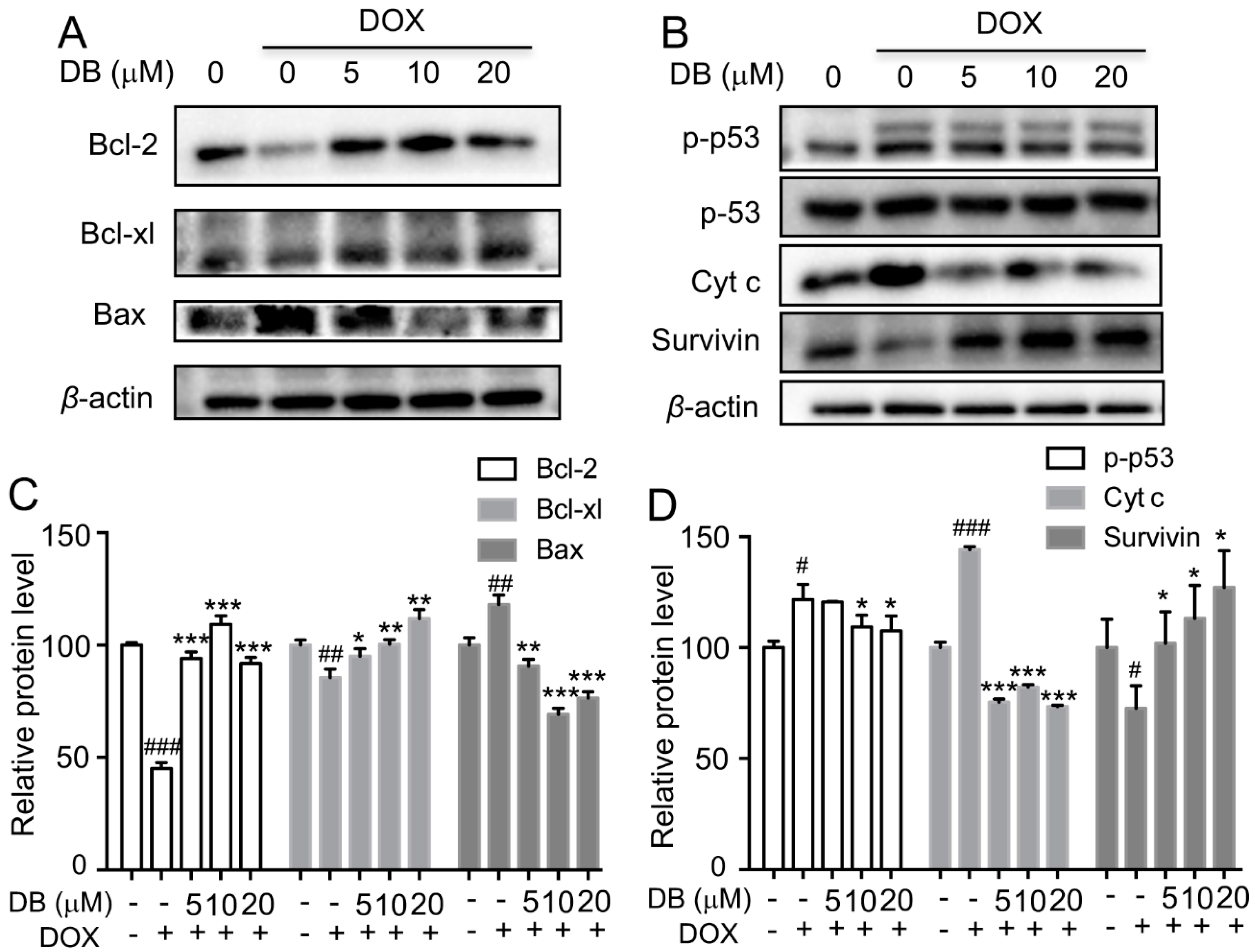
2.3. DB Protects from DOX-Induced Apoptosis by Modulation Bcl-2 Family Proteins
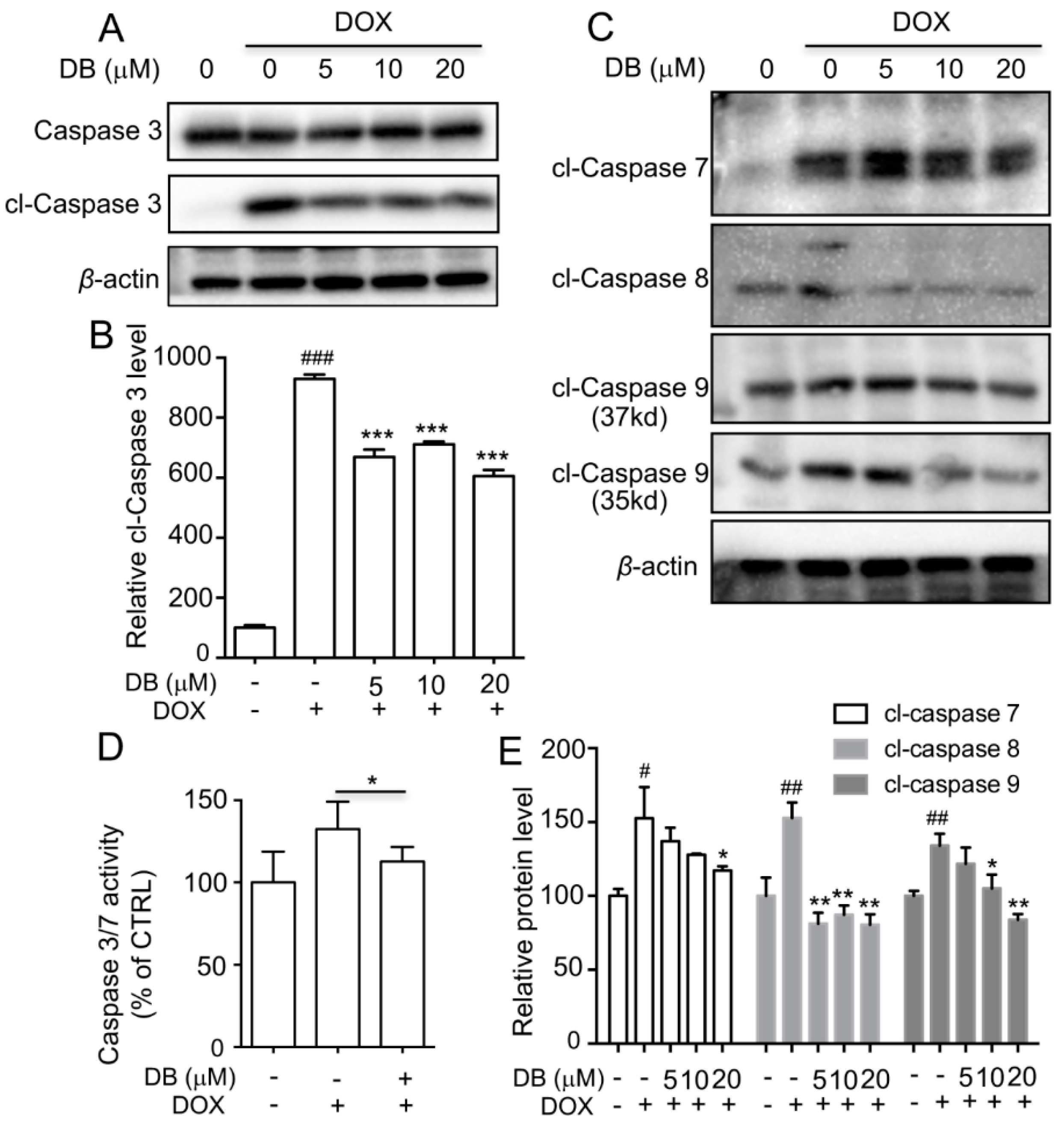
2.4. DB Protects from DOX-Induced Apoptosis by Modulation of Caspases
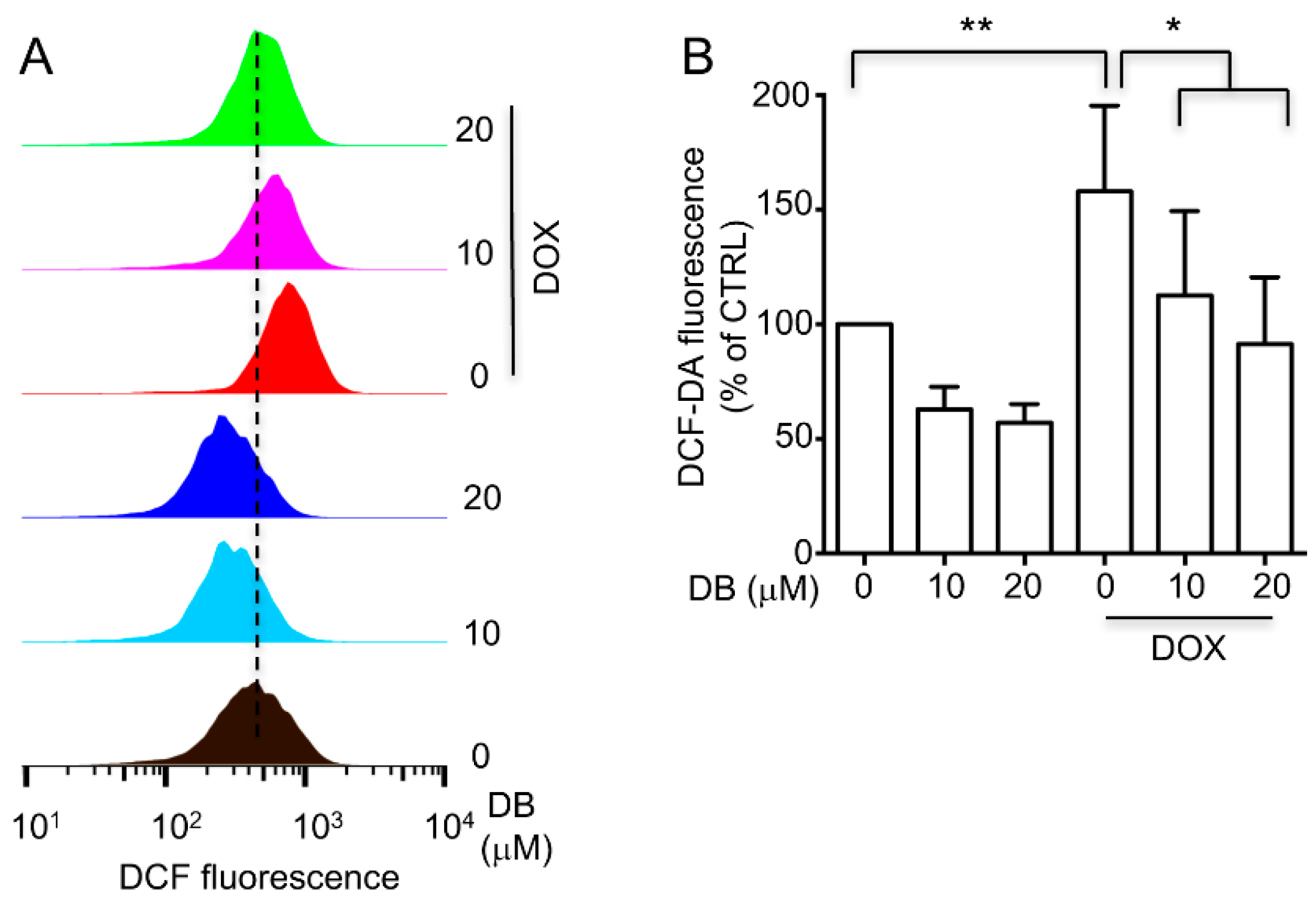
2.5. DB Inhibited DOX-Induced Oxidative Stress
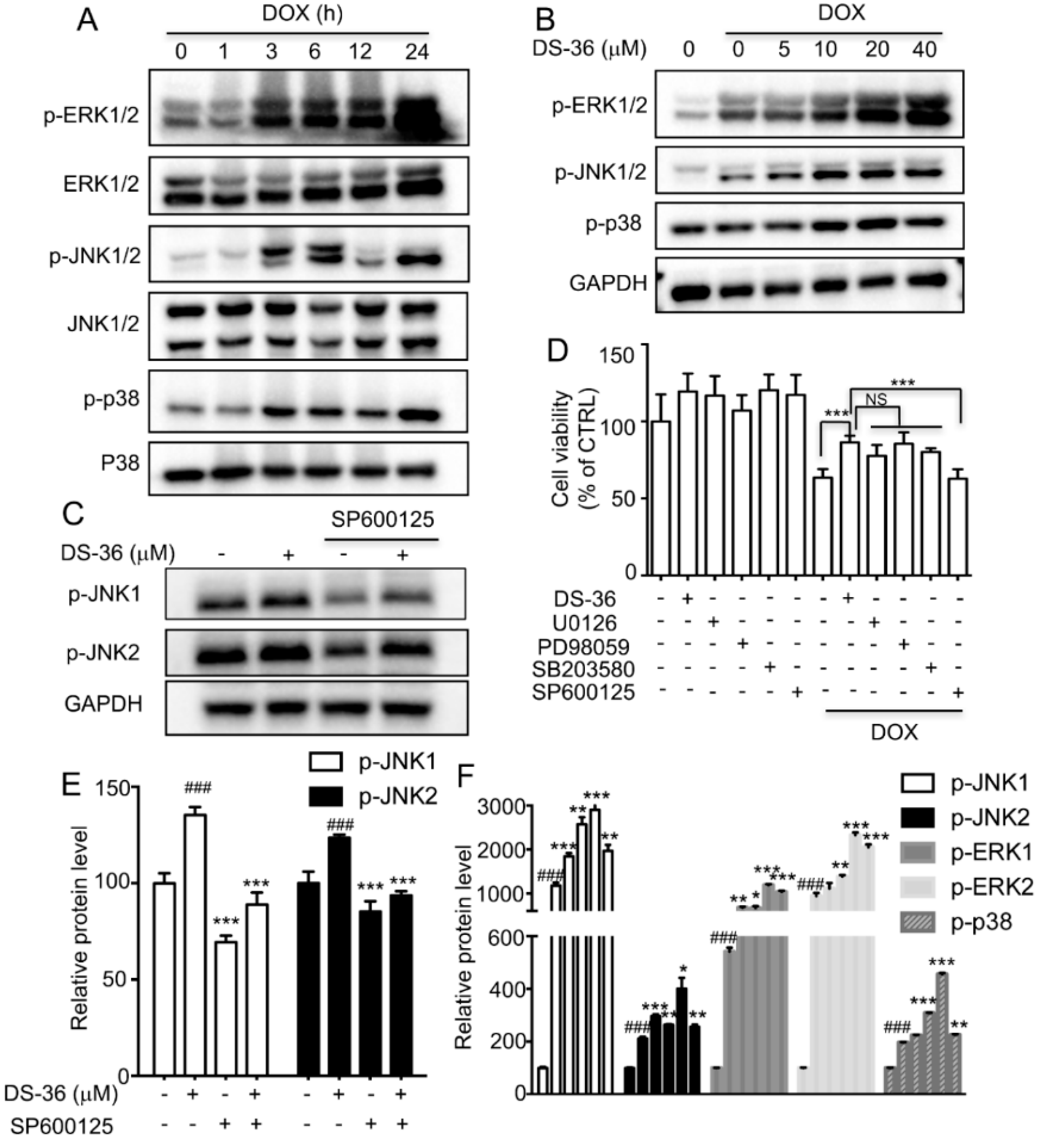
2.6. DB Protected from DOX-Induced Apoptosis by Activating JNK1/2
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. MTT Assay
4.4. Detection of Intracellular ROS
4.5. Mitochondria Membrane Potential (MMP) Evaluation
4.6. Western Blotting
4.7. Caspase Activity Measurement
4.8. Apoptosis Assay
4.9. TUNEL Staining
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. Caesius. Biotechnol. Bioeng. 2000, 67, 704–713, reprinted from 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Shenkenberg, T.D.; Von Hoff, D.D. Mitoxantrone: A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1986, 105, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Ludke, A.R.; Al-Shudiefat, A.A.; Dhingra, S.; Jassal, D.S.; Singal, P.K. A concise description of cardioprotective strategies in doxorubicin-induced cardiotoxicity. Can. J. Physiol. Pharmacol. 2009, 87, 756–763. [Google Scholar] [PubMed]
- Kane, R.C.; McGuinn, W.D., Jr.; Dagher, R.; Justice, R.; Pazdur, R. Dexrazoxane (Totect™): FDA review and approval for the treatment of accidental extravasation following intravenous anthracycline chemotherapy. Oncologist 2008, 13, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, F.; Dupuis, L.L.; Alexander, S.; Gupta, A.; Mertens, L.; Nathan, P.C. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2016, 108, djv357. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Peng, X.; Pentassuglia, L.; Lim, C.C.; Sawyer, D.B. Molecular and cellular mechanisms of anthracycline cardiotoxicity. Cardiovasc. Toxicol. 2007, 7, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.X.; Khechaduri, A.; Prasad, S.V.N.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarska-Schlattner, M.; Zaugg, M.; da Silva, R.; Lucchinetti, E.; Schaub, M.C.; Wallimann, T.; Schlattner, U. Acute toxicity of doxorubicin on isolated perfused heart: Response of kinases regulating energy supply. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H37–H47. [Google Scholar] [CrossRef] [PubMed]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of chemotherapeutic agents: Incidence, treatment and prevention. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef] [PubMed]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-induced cardiotoxicity: Overview of the roles of oxidative stress. Oxid. Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Shi, J.; Li, Y.J.; Wei, L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch. Immunol. Ther. Exp. 2009, 57, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.Y.; Tan, B.K.; Zhu, Y.Z. Salvia miltiorrhiza and ischemic diseases. Acta Pharmacol. Sin. 2000, 21, 1089–1094. [Google Scholar] [PubMed]
- Zhou, R.; He, L.F.; Li, Y.J.; Shen, Y.; Chao, R.B.; Du, J.R. Cardioprotective effect of water and ethanol extract of Salvia miltiorrhiza in an experimental model of myocardial infarction. J. Ethnopharmacol. 2012, 139, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.W.; Sun, W.; Zhao, J.P.; Wu, X.X.; Lu, J.J.; Chen, X.P.; Xu, Q.M.; Khan, I.A.; Yang, S.L. Tanshinones and diethyl blechnics with anti-inflammatory and anti-cancer activities from Salvia miltiorrhiza bunge (danshen). Sci. Rep. 2016, 6, 33720. [Google Scholar] [CrossRef] [PubMed]
- Cvetkokic, R.S.; Scott, L.J. Dexrazoxane—A review of its use for cardioprotection during anthracycline chemotherapy. Drugs 2005, 65, 1005–1024. [Google Scholar]
- Jiang, B.H.; Zhang, L.; Wang, Y.C.; Li, M.; Wu, W.Y.; Guan, S.H.; Liu, X.; Yang, M.; Wang, J.C.; Guo, D.A. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food Chem. Toxicol. 2009, 47, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.H.; Chen, L.; Lu, Q.W.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G.Y. Quercetin attenuates doxorubicin cardiotoxicity by modulating BMI-1 expression. Br. J. Pharmacol. 2014, 171, 4440–4454. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.M.; Al-Harthi, S.E.; AlArabi, O.M.; Elshal, M.F.; Ramadan, W.S.; Alaama, M.N.; Al-Kreathy, H.M.; Damanhouri, Z.A.; Osman, O.H. Chemosensetizing and cardioprotective effects of resveratrol in doxorubicin-treated animals. Cancer Cell Int. 2013, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Green, P.S.; Leeuwenburgh, C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. BBA-Mol. Basis Dis. 2002, 1588, 94–101. [Google Scholar] [CrossRef]
- Nithipongvanitch, R.; Ittarat, W.; Cole, M.P.; Tangpong, J.; Clair, D.K.; Oberley, T.D. Mitochondrial and nuclear p53 localization in cardiomyocytes: Redox modulation by doxorubicin (adriamycin)? Antioxid. Redox Signal. 2007, 9, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.C.; Phaneuf, S.L.; Dirks, A.J.; Phillips, T.; Leeuwenburgh, C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002, 62, 4592–4598. [Google Scholar] [PubMed]
- Ghibu, S.; Delemasure, S.; Richard, C.; Guilland, J.C.; Martin, L.; Gambert, S.; Rochette, L.; Vergely, C. General oxidative stress during doxorubicin-induced cardiotoxicity in rats: Absence of cardioprotection and low antioxidant efficiency of α-lipoic acid. Biochimie 2012, 94, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Alshabanah, O.A.; Hafez, M.M.; Al-Harbi, M.M.; Hassan, Z.K.; Al Rejaie, S.S.; Asiri, Y.A.; Sayed-Ahmed, M.M. Doxorubicin toxicity can be ameliorated during antioxidant l-carnitine supplementation. Oxid. Med. Cell. Longev. 2010, 3, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; El-Azem, N.; Pamplona, R.; Ramirez-Tortosa, C.; Pulido-Moran, M.; Vera-Ramirez, L.; Quiles, J.L.; Sanchez-Rovira, P.; Naudi, A.; Portero-Otin, M.; et al. Hydroxytyrosol ameliorates oxidative stress and mitochondrial dysfunction in doxorubicin-induced cardiotoxicity in rats with breast cancer. Biochem. Pharmacol. 2014, 90, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Konorev, E.A.; Kotamraju, S.; Zhao, H.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Paradoxical effects of metalloporphyrins on doxorubicin-induced apoptosis: Scavenging of reactive oxygen species versus induction of heme oxygenase-1. Free Radic. Biol. Med. 2002, 33, 988–997. [Google Scholar] [CrossRef]
- Hong, H.J.; Liu, J.C.; Chen, P.Y.; Chen, J.J.; Chan, P.; Cheng, T.H. Tanshinone IIA prevents doxorubicin-induced cardiomyocyte apoptosis through AKT-dependent pathway. Int. J. Cardiol. 2012, 157, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.J.; Chan, J.Y.W.; Shan, L.C.; Cui, G.Z.; Cui, Q.B.; Wang, Y.F.; Li, J.J.; Chen, H.X.; Zhang, Q.W.; et al. A novel danshensu derivative prevents cardiac dysfunction and improves the chemotherapeutic efficacy of doxorubicin in breast cancer cells. J. Cell. Biochem. 2016, 117, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Adachi, S.; Ito, H.; Hirao, K.; Isobe, M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell 2008, 7, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, P.; Barisione, C.; Lazzarini, E.; Garuti, A.; Bezante, G.P.; Canepa, M.; Spallarossa, P.; Tocchetti, C.G.; Bollini, S.; Brunelli, C.; et al. Testosterone antagonizes doxorubicin-induced senescence of cardiomyocytes. J. Am. Heart Assoc. 2016, 5, e002383. [Google Scholar] [CrossRef] [PubMed]
- Adderley, S.R.; Fitzgerald, D.J. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J. Biol. Chem. 1999, 274, 5038–5046. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Varadharaj, S.; Ganesan, L.P.; Shobha, J.C.; Naidu, M.U.; Parinandi, N.L.; Tridandapani, S.; Kutala, V.K.; Kuppusamy, P. C-phycocyanin protects against ischemia-reperfusion injury of heart through involvement of p38 MAPK and ERK signaling. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H2136–H2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Force, T.; Bonventre, J.V. Growth factors and mitogen-activated protein kinases. Hypertension 1998, 31, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. The protective role of arjunolic acid against doxorubicin induced intracellular ros dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials 2011, 32, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Mantawy, E.M.; Esmat, A.; El-Bakly, W.M.; Eldin, R.A.S.; El-Demerdash, E. Mechanistic clues to the protective effect of chrysin against doxorubicin-induced cardiomyopathy: Plausible roles of p53, MAPK and AKT pathways. Sci. Rep. 2017, 7, 4795. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.A.; Liang, Q.R.; Bueno, O.; Huang, Y.; Lackey, T.; Klevitsky, R.; Hewett, T.E.; Molkentin, J.D. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J. Biol. Chem. 2005, 280, 32602–32608. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.Q.; Wang, X.R.; Liu, M.; Xu, F.F.; Liu, X.H. Myofibrillogenesis regulator-1 attenuated hypoxia/reoxygenation-induced apoptosis by inhibiting the PERK/NRF2 pathway in neonatal rat cardiomyocytes. Apoptosis 2015, 20, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Bao, J.; Lin, W.; Gao, H.; Zhao, W.; Zhang, Q.; Leung, C.H.; Ma, D.L.; Lu, J.; Chen, X. 2-Methoxy-6-acetyl-7-methyljuglone (MAM), a natural naphthoquinone, induces NO-dependent apoptosis and necroptosis by H2O2-dependent JNK activation in cancer cells. Free Radic. Biol. Med. 2016, 92, 61–77. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Gao, H.; Wu, C.; Xu, Q.-M.; Lu, J.-J.; Chen, X. Diethyl Blechnic, a Novel Natural Product Isolated from Salvia miltiorrhiza Bunge, Inhibits Doxorubicin-Induced Apoptosis by Inhibiting ROS and Activating JNK1/2. Int. J. Mol. Sci. 2018, 19, 1809. https://doi.org/10.3390/ijms19061809
Yu J, Gao H, Wu C, Xu Q-M, Lu J-J, Chen X. Diethyl Blechnic, a Novel Natural Product Isolated from Salvia miltiorrhiza Bunge, Inhibits Doxorubicin-Induced Apoptosis by Inhibiting ROS and Activating JNK1/2. International Journal of Molecular Sciences. 2018; 19(6):1809. https://doi.org/10.3390/ijms19061809
Chicago/Turabian StyleYu, Jie, Hongwei Gao, Chuanhong Wu, Qiong-Ming Xu, Jin-Jian Lu, and Xiuping Chen. 2018. "Diethyl Blechnic, a Novel Natural Product Isolated from Salvia miltiorrhiza Bunge, Inhibits Doxorubicin-Induced Apoptosis by Inhibiting ROS and Activating JNK1/2" International Journal of Molecular Sciences 19, no. 6: 1809. https://doi.org/10.3390/ijms19061809