Identification of an Actionable Mutation of KIT in a Case of Extraskeletal Myxoid Chondrosarcoma

 , , , , , , ,
, , , , , , ,
Abstract
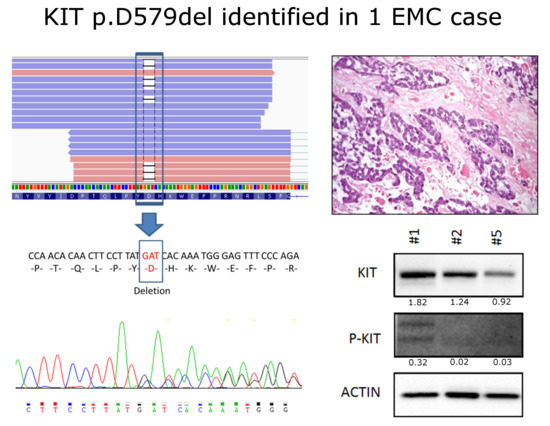
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
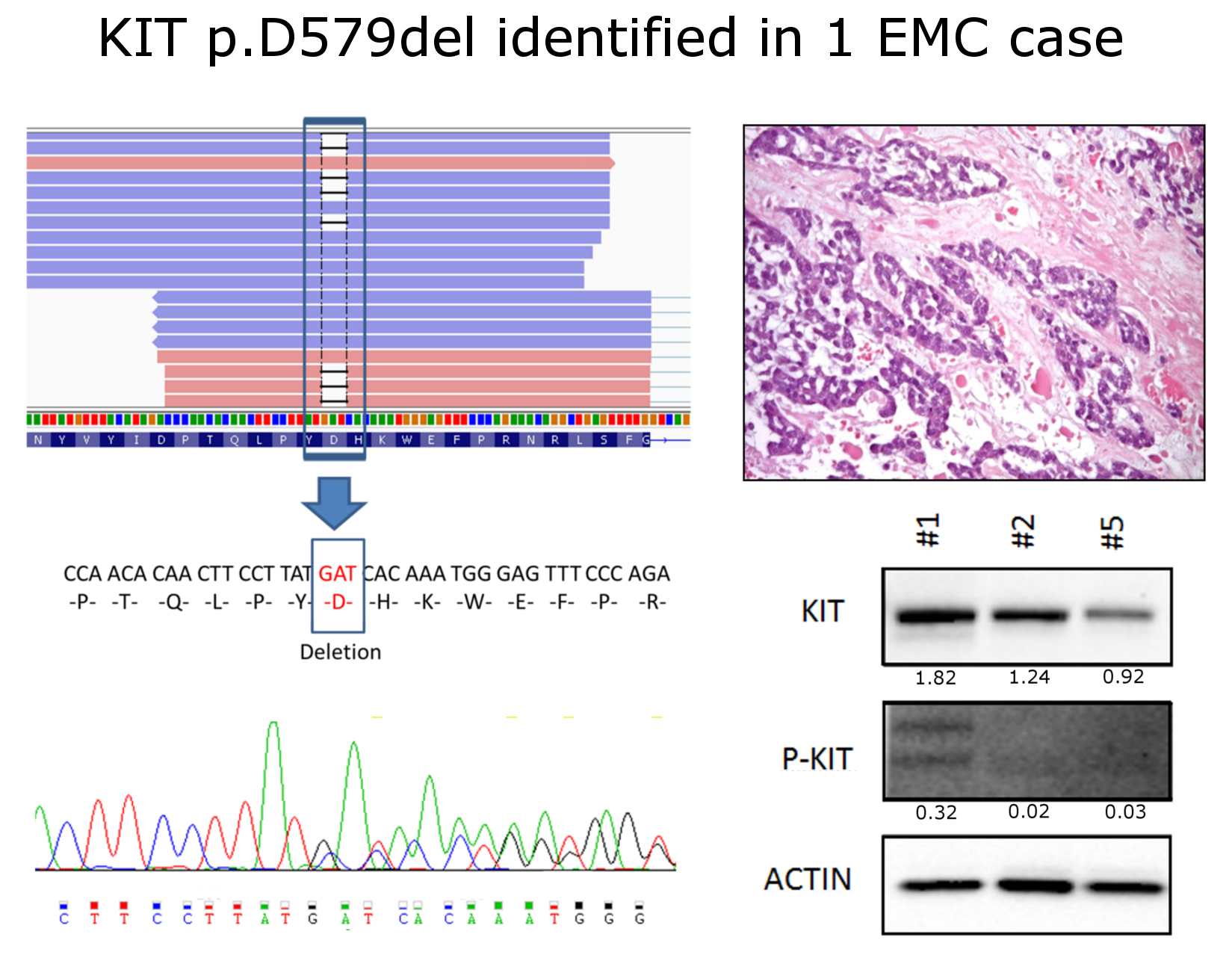
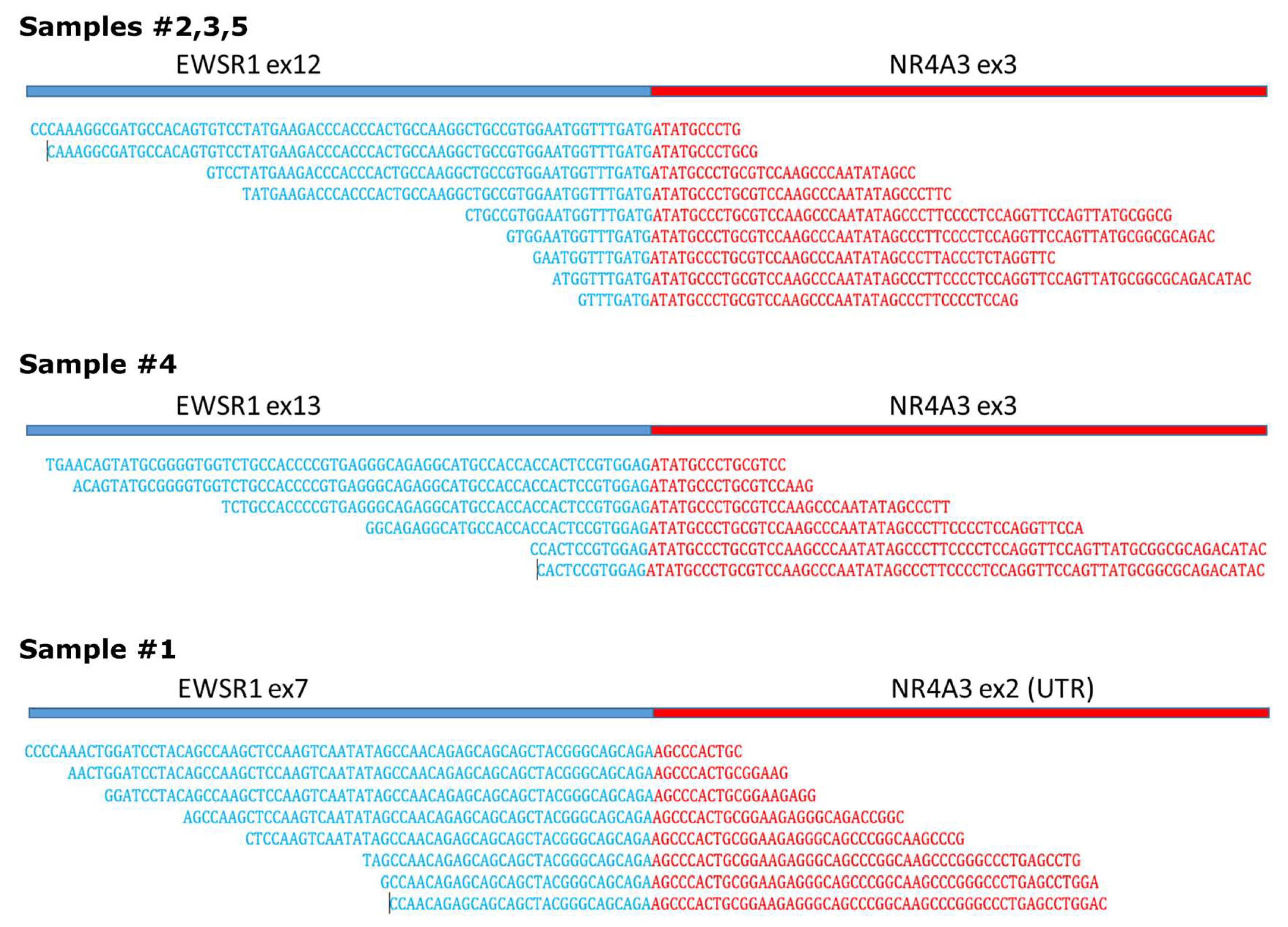
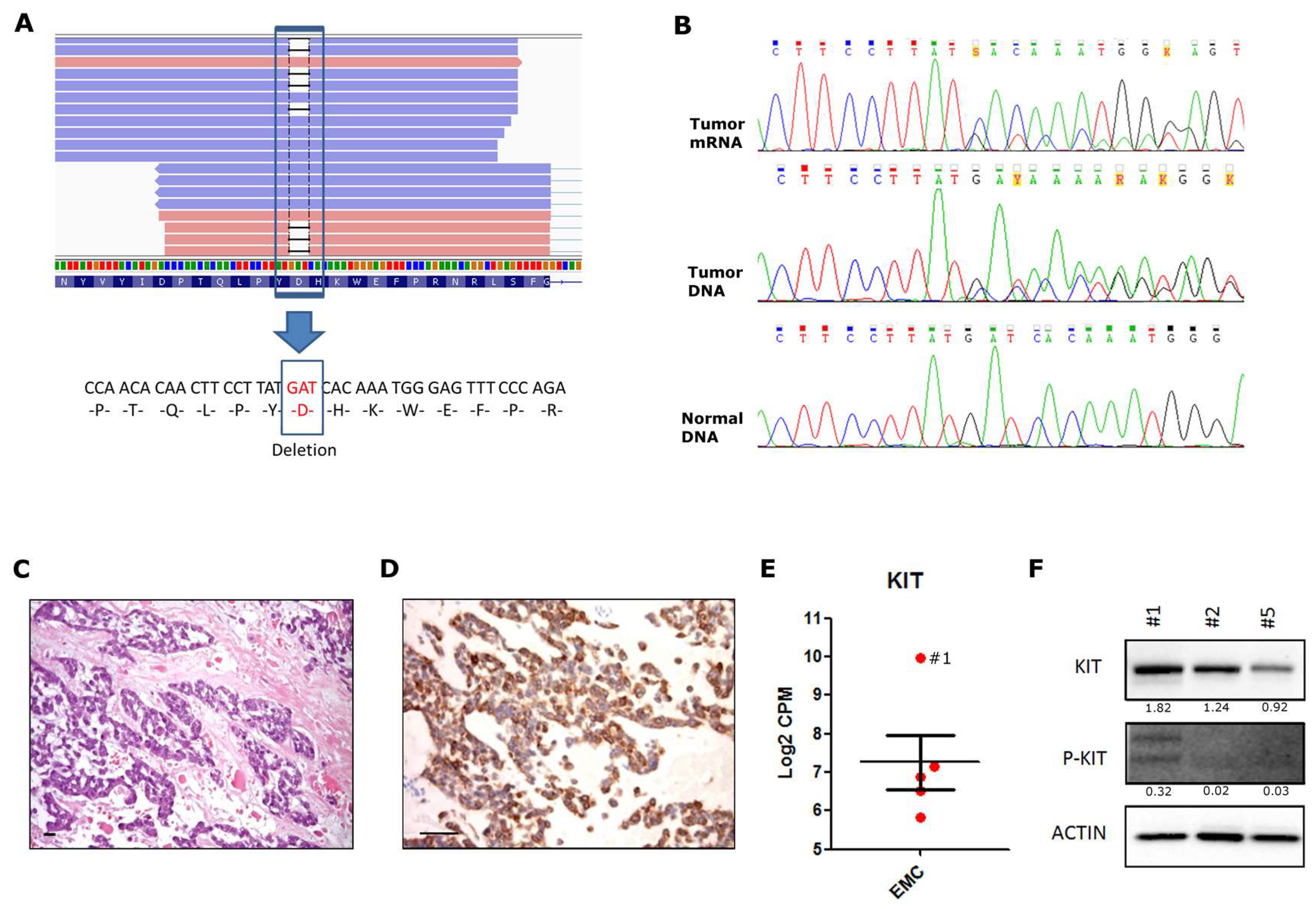
2. Results and Discussion
3. Materials and Methods
3.1. Samples
3.2. Whole Transcriptome Sequencing
3.3. Bioinformatic Analysis
3.4. Sanger Sequencing
3.5. Immunohistochemistry
3.6. Western Blot
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Fletcher, C.D.M.; Unni, K.K.; Mertens, F. (Eds.) World Health Organization (WHO) Classification of Tumours—Pathology and Genetics of Tumours of Soft Tissue and Bone; IARC Press: Lyon, France, 2013. [Google Scholar]
- Hinrichs, S.H.; Jaramillo, M.A.; Gumerlock, P.H.; Gardner, M.B.; Lewis, J.P.; Freeman, A.E. Myxoid chondrosarcoma with a translocation involving chromosomes 9 and 22. Cancer Genet. Cytogenet. 1985, 14, 219–226. [Google Scholar] [CrossRef]
- Flucke, U.; Tops, B.B.; Verdijk, M.A.; van Cleef, P.J.; van Zwam, P.H.; Slootweg, P.J.; Bovée, J.V.; Riedl, R.G.; Creytens, D.H.; Suurmeijer, A.J.; et al. NR4A3 rearrangement reliably distinguishes between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch. 2012, 460, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, Y.; Zucman, J.; Stenman, G.; Kindblom, L.G.; Knight, J.; Turc-Carel, C.; Dockhorn-Dworniczak, B.; Mandahl, N.; Desmaze, C.; Peter, M.; et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum. Mol. Genet. 1995, 4, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Domanski, H.A.; Brosjö, O.; Heim, S.; Bjerkehagen, B.; Sciot, R.; Dal Cin, P.; Fletcher, J.A.; et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoidchondrosarcoma. Genes Chromosomes Cancer 2002, 35, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; Zhang, L.; Sung, Y.S.; Singer, S.; Antonescu, C.R. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum. Pathol. 2014, 45, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Urbini, M.; Astolfi, A.; Pantaleo, M.A.; Serravalle, S.; Dei Tos, A.P.; Picci, P.; Indio, V.; Sbaraglia, M.; Benini, S.; Righi, A.; et al. HSPA8 as a novel fusion partner of NR4A3 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2017, 56, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Fujiwara, T.; Beppu, Y.; Chuman, H.; Yoshida, A.; Kawano, H.; Kawai, A. Extraskeletal myxoid chondrosarcoma: A review of 23 patients treated at a single referral center with long-term follow-up. Arch. Orthop. Trauma Surg. 2012, 132, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.D.; Popat, S.; Bhuchar, G.; D’Adamo, D.R.; Keohan, M.L.; Fisher, C.; Antonescu, C.R.; Singer, S.; Brennan, M.F.; Judson, I.; et al. Extraskeletal myxoid chondrosarcoma: A retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 2008, 113, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Dagrada, G.P.; Sanfilippo, R.; Negri, T.; Vittimberga, I.; Ferrari, S.; Grosso, F.; Apice, G.; Tricomi, M.; Colombo, C.; et al. Anthracycline-based chemotherapy in extraskeletal myxoid chondrosarcoma: A retrospective study. Clin. Sarcoma Res. 2013, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Dagrada, G.P.; Morosi, C.; Negri, T.; Romanini, A.; Pilotti, S.; Gronchi, A.; Casali, P.G. Extraskeletal myxoid chondrosarcoma: Tumor response to sunitinib. Clin. Sarcoma Res. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Pantaleo, M.A.; Astolfi, A.; Dagrada, G.P.; Negri, T.; Dei Tos, A.P.; Indio, V.; Morosi, C.; Gronchi, A.; Colombo, C.; et al. Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur. J. Cancer 2014, 50, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Wu, Y.M.; Robinson, D.; Schuetze, S.M.; Baker, L.H.; Athanikar, J.; Cao, X.; Kunju, L.P.; Chinnaiyan, A.M.; Chugh, R. Next generation sequencing of extraskeletal myxoid chondrosarcoma. Oncotarget 2017, 8, 21770–21777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, M.; Isozaki, K.; Hirota, S.; Miyagawa, J.; Hase-Sawada, N.; Taniguchi, M.; Nishida, T.; Kanayama, S.; Kitamura, Y.; Shinomura, Y.; et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998, 115, 1090–1095. [Google Scholar] [CrossRef]
- Dieter, S.M.; Heining, C.; Agaimy, A.; Huebschmann, D.; Bonekamp, D.; Hutter, B.; Ehrenberg, K.R.; Fröhlich, M.; Schlesner, M.; Scholl, C.; et al. Mutant KIT as imatinib-sensitive target in metastatic sinonasal carcinoma. Ann. Oncol. 2017, 28, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S.; Govindan, R.; Javidan-Nejad, C.; Pfeifer, J.D.; Cottrell, C.E. Stabilization of disease after targeted therapy in a thymic carcinoma with KIT mutation detected by clinical next-generation sequencing. J. Thorac. Oncol. 2014, 9, e12–e16. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbini, M.; Indio, V.; Astolfi, A.; Tarantino, G.; Renne, S.L.; Pilotti, S.; Dei Tos, A.P.; Maestro, R.; Collini, P.; Nannini, M.; et al. Identification of an Actionable Mutation of KIT in a Case of Extraskeletal Myxoid Chondrosarcoma. Int. J. Mol. Sci. 2018, 19, 1855. https://doi.org/10.3390/ijms19071855
Urbini M, Indio V, Astolfi A, Tarantino G, Renne SL, Pilotti S, Dei Tos AP, Maestro R, Collini P, Nannini M, et al. Identification of an Actionable Mutation of KIT in a Case of Extraskeletal Myxoid Chondrosarcoma. International Journal of Molecular Sciences. 2018; 19(7):1855. https://doi.org/10.3390/ijms19071855
Chicago/Turabian StyleUrbini, Milena, Valentina Indio, Annalisa Astolfi, Giuseppe Tarantino, Salvatore Lorenzo Renne, Silvana Pilotti, Angelo Paolo Dei Tos, Roberta Maestro, Paola Collini, Margherita Nannini, and et al. 2018. "Identification of an Actionable Mutation of KIT in a Case of Extraskeletal Myxoid Chondrosarcoma" International Journal of Molecular Sciences 19, no. 7: 1855. https://doi.org/10.3390/ijms19071855