Phosphokinome Analysis of Barth Syndrome Lymphoblasts Identify Novel Targets in the Pathophysiology of the Disease

 , ,
, ,
Abstract
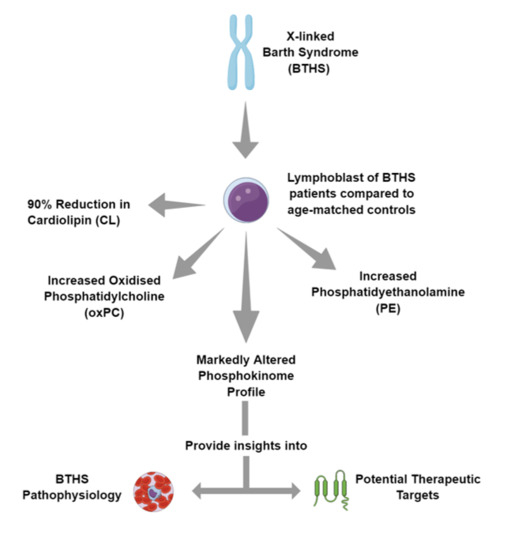
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetler, K.Y. Chapter 6 Polyglycerophospholipids: Phosphatidylglycerol, diphosphatidylglycerol and bis (monoacylglycero) phosphate. New Compr. Biochem. 1982, 4, 215–261. [Google Scholar]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta 2017, 1862, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini-Chohan, H.K.; Mitchell, R.W.; Vaz, F.M.; Zelinski, T.; Hatch, G.M. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders. J. Lipid Res. 2012, 53, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Hatch, G.M. Cardiolipin biosynthesis in the isolated heart. Biochem. J. 1994, 297 (Pt 1), 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparagna, G.C.; Johnson, C.A.; McCune, S.A.; Moore, R.L.; Murphy, R.C. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J. Lipid Res. 2005, 46, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Kelley, R.I.; Blanck, T.J.J.; Schlame, M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol Chem. 2003, 278, 51380–51385. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The Enzymatic Function of Tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.; Guerra, G.; Padrão, A.I.; Melo, T.; Vitorino, R.; Duarte, J.A. Lipidomic characterization of streptozotocin-induced heart mitochondrial dysfunction. Mitochondrion 2013, 13, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Hauff, K.D.; Hatch, G.M. Cardiolipin metabolism and Barth Syndrome. Prog. Lipid Res. 2006, 45, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Saric, A.; Andreau, K.; Armand, A.-S.; Møller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, G.J.; McMaster, C.R. Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem. Phys. Lipids 2015, 193, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vreken, P.; Valianpour, F.; Nijtmans, L.G.; Grivell, L.A.; Plecko, B.; Wanders, R.J. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 2000, 279, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Shanske, S.; Doty, S.; König, T.; Sculco, T.; DiMauro, S. Microanalysis of cardiolipin in small biopsies including skeletal muscle from patients with mitochondrial disease. J. Lipid Res. 1999, 40, 1585–1592. [Google Scholar] [PubMed]
- Schlame, M.; Towbin, J.A.; Heerdt, P.M.; Jehle, R.; DiMauro, S.; Blanck, T.J.J. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 2002, 51, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Brandner, K.; Mick, D.U.; Frazier, A.E.; Taylor, R.D.; Meisinger, C.; Rehling, P. Taz1, an Outer Mitochondrial Membrane Protein, Affects Stability and Assembly of Inner Membrane Protein Complexes: Implications for Barth Syndrome. Mol. Biol. Cell. 2005, 16, 5202–5214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.-P.; Landes, T. Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 2013, 1832, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.A.; Mejia, E.M.; Mitchell, R.W.; Choy, P.C.; Sparagna, G.C.; Hatch, G.M. Human Trifunctional Protein Alpha Links Cardiolipin Remodeling to Beta-Oxidation. PLoS ONE. 2012, 7, e48628. [Google Scholar] [CrossRef] [PubMed]
- Mejia, E.M.; Zegallai, H.; Bouchard, E.D.; Banerji, V.; Ravandi, A.; Hatch, G.M. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth Syndrome lymphoblasts. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sutachan, J.J.; Plesken, H.; Kelley, R.I.; Schlame, M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Investig. 2005, 85, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; He, Q.; Greenberg, M.L. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 2008, 68, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Thompson, M.N.; Greenberg, M.L. Synthetic Lethal Interaction of the Mitochondrial Phosphatidylethanolamine and Cardiolipin Biosynthetic Pathways in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 35410–35416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, L.K.; Mejia, E.M.; Vandel, M.; Sparagna, G.C.; Claypool, S.M.; Dyck-Chan, L. Impaired Cardiolipin Biosynthesis Prevents Hepatic Steatosis and Diet-Induced Obesity. Diabetes 2016, 65, 3289–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.; Thompson, R.; Vernon, H. Barth Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK247162/ (accessed on 13 May 2018).
- Mejia, E.M.; Zinko, J.C.; Hauff, K.D.; Xu, F.Y.; Ravandi, A.; Hatch, G.M. Glucose Uptake and Triacylglycerol Synthesis Are Increased in Barth Syndrome Lymphoblasts. Lipids 2017, 52, 161–165. [Google Scholar] [CrossRef] [PubMed]
- He, Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H210–H216. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, M.; Harris, N.; Han, X. Tafazzin knockdown interrupts cell cycle progression in cultured neonatal ventricular fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1332–H1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeulder, B.; Zarrinpashneh, E.; Ginion, A.; Viollet, B.; Hue, L.; Rider, M.H.; Vanoverschelde, J.L.; Beauloye, C.; Horman, S.; Bertrand, L. Differential regulation of eEF2 and p70S6K by AMPKalpha2 in heart. Biochim. Biophys. Acta 2013, 1832, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baruzzi, A.; Caveggion, E.; Berton, G. Regulation of phagocyte migration and recruitment by Src-family kinases. Cell. Mol. Life Sci. 2008, 65, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Liao, J.; Liu, X.; Wang, P.; Liu, J.; Hou, W. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell Cycle 2011, 10, 1411–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkhabi, M.; Razavi, S.M.; Salari, A. Global transcriptomic analysis of induced cardiomyocytes predicts novel regulators for direct cardiac reprogramming. J. Cell Commun. Signal. 2017, 11, 193–204. [Google Scholar] [CrossRef] [PubMed]
- De Mesquita, D.D.; Zhan, Q.; Crossley, L.; Badwey, J.A. p90-RSK and Akt may promote rapid phosphorylation/inactivation of glycogen synthase kinase 3 in chemoattractant-stimulated neutrophils. FEBS Lett. 2001, 502, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Kawagoe, T.; Sato, S.; Jung, A.; Yamamoto, M.; Matsui, K.; Kato, H. Essential role of IRAK-4 protein and its kinase activity in Toll-like receptor–mediated immune responses but not in TCR signaling. J. Exp. Med. 2007, 204, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Chen, J. Mammalian target of rapamycin (mTOR) signaling network in skeletal myogenesis. J. Biol. Chem. 2012, 287, 43928–43935. [Google Scholar] [CrossRef] [PubMed]
- Mala, J.G.S.; Takeuchi, S. Molecular cloning of OSP94: A significant biomarker protein of hypertensive human heart and a member of HSP110 family. Mol. Biotechnol. 2009, 42, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Shorofsky, M.; Maguy, A.; Nattel, S. Consequences of Atrial or Ventricular Tachypacing on the Heat Shock Proteins (HSP) level of Expression and Phosphorylation. McGill J. Med. 2009, 12, 34. [Google Scholar] [PubMed]
- Dohke, T.; Wada, A.; Isono, T.; Fujii, M.; Yamamoto, T.; Tsutamoto, T. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J. Card. Fail. 2006, 12, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yung, A.; Covarrubias, M.; Radice, G.L. Cortactin Is Required for N-cadherin Regulation of Kv1.5 Channel Function. J. Biol. Chem. 2011, 286, 20478–20489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowa, G.; Pypaert, M.; Fulton, D.; Sessa, W.C. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc. Natl. Acad. Sci. USA 2003, 100, 6511–6516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardt, S.E.; Tomita, H.; Katus, H.A.; Sadoshima, J. Phosphorylation of eukaryotic translation initiation factor 2Bepsilon by glycogen synthase kinase-3beta regulates beta-adrenergic cardiac myocyte hypertrophy. Circ. Res. 2004, 94, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Bouayad, D.; Pederzoli-Ribeil, M.; Mocek, J.; Candalh, C.; Arlet, J.-B.; Hermine, O. Nuclear-to-cytoplasmic relocalization of the proliferating cell nuclear antigen (PCNA) during differentiation involves a chromosome region maintenance 1 (CRM1)-dependent export and is a prerequisite for PCNA antiapoptotic activity in mature neutrophils. J. Biol. Chem. 2012, 287, 33812–33825. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.L.; Cui, M.; Hernandez, J.B.; Weist, B.M.; Andersen, H.-M.; Zhang, X. Modulation of DRAK2 autophosphorylation by antigen receptor signaling in primary lymphocytes. J. Biol. Chem. 2007, 282, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, M.; Tschan, M.P.; Britschgi, C.; Britschgi, A.; Hügli, B.; Grob, T.J. The death-associated protein kinase 2 is up-regulated during normal myeloid differentiation and enhances neutrophil maturation in myeloid leukemic cells. J. Leukoc. Biol. 2007, 81, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.; Nebreda, A.R. Regulation of Cdc25C activity during the meiotic G2/M. transition. Cell Cycle 2004, 3, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Berry, R. The Role of L(u)ck in T Cell Triggering. Sci. Signal. 2011, 4, jc2. [Google Scholar] [CrossRef] [PubMed]
- Haigh, J.J.; Ema, M.; Haigh, K.; Gertsenstein, M.; Greer, P.; Rossant, J. Activated Fps/Fes partially rescues the in vivo developmental potential of Flk1-deficient vascular progenitor cells. Blood 2004, 103, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Sanyal, S.; Mukhopadhyay, D. Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 2001, 276, 32714–32719. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Kobayashi, K.; Mikoshiba, K.; Nakajima, K. Regulation of cortical neuron migration by the Reelin signaling pathway. Neurochem. Res. 2011, 36, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ochalski, P.G.; Tran, T.S.; Sahir, N.; Schubert, M.; Pramatarova, A. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J. Cell Sci. 2004, 117, 4527–4536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.; Su, Q.; Xi, P.; Han, S.; Li, J. Monocular deprivation delays the dynamic changes of phosphorylated synapsin Ia/b at site-1 in contralateral visual cortex of juvenile mice. Neurochem. Res. 2015, 40, 524–530. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgis, J.A.; Jaffe, H.; Moreira, J.E.; Carlotti, C.G.; Leite, J.P.; Pant, H.C. Phosphoproteomic analysis of synaptosomes from human cerebral cortex. J. Proteome Res. 2005, 4, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Philipp, M.; Evron, T.; Caron, M.G. The role of arrestins in development. Prog. Mol. Biol. Transl. Sci. 2013, 118, 225–242. [Google Scholar] [PubMed]
- Balabanian, K.; Levoye, A.; Klemm, L.; Lagane, B.; Hermine, O.; Harriague, J. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J. Clin. Investig. 2008, 118, 1074–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valsecchi, F.; Konrad, C.; Manfredi, G. Role of soluble adenylyl cyclase in mitochondria. Biochim. Biophys. Acta 2014, 1842, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.J.; Nakamoto, M.; Bergemann, A.D.; Flanagan, J.G. Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell 1995, 82, 371–381. [Google Scholar] [CrossRef]
- Beggs, J.E.; Tian, S.; Jones, G.G.; Xie, J.; Iadevaia, V.; Jenei, V. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem. J. 2015, 467, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.C.; Geiger, P.C.; Han, D.-H.; Jones, T.E.; Holloszy, J.O. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007, 282, 18793–18799. [Google Scholar] [CrossRef] [PubMed]
- Thévenin, A.F.; Zony, C.L.; Bahnson, B.J.; Colman, R.F. Activation by phosphorylation and purification of human c-Jun N-terminal kinase (JNK) isoforms in milligram amounts. Protein Expr. Purif. 2011, 75, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, J.L.; Kulkarni, R.; Randis, T.M.; Soman, S.; Kikuchi, A.; Yin, Y. Phosphatase-dependent regulation of epithelial mitogen-activated protein kinase responses to toxin-induced membrane pores. PLoS ONE 2009, 4, e8076. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zhong, H.; Hayden, M.S.; Ghosh, S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008, 22, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.; Yoshida, K.; Ushiyama, M.; Yamaguchi, T.; Iwashita, K.; Futagawa, T. The small heat shock protein alphaB-crystallin inhibits differentiation-induced caspase 3 activation and myogenic differentiation. Biol. Pharm. Bull. 2006, 29, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Okamoto, K.; Nakayama, H.; Isobe, T.; Kato, K. Phosphorylation of alphaB-crystallin in response to various types of stress. J. Biol. Chem. 1997, 272, 29934–29941. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.A.; Mewburn, J.D.; Truesdell, P.; Greer, P.A. The Fps/Fes kinase regulates leucocyte recruitment and extravasation during inflammation. Immunology 2007, 122, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khajah, M.; Andonegui, G.; Chan, R.; Craig, A.W.; Greer, P.A.; McCafferty, D.-M. Fer kinase limits neutrophil chemotaxis toward end target chemoattractants. J. Immunol. 2013, 190, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, A.; Leibundgut, G.; Hung, M.-Y.; Patel, M.; Hutchins, P.M.; Murphy, R.C. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. J. Am. Coll. Cardiol. 2014, 63, 1961–1971. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Phospholipid | BTHS | Control |
|---|---|---|
| PC | 69.66615 | 69.75617 |
| OxPC | 0.392307 | 0.264211 |
| LPC | 0.161851 | 0.332797 |
| SM | 4.488527 | 4.887775 |
| CL | 0.030898 | 0.847094 |
| PS | 12.26267 | 14.17647 |
| PG | 3.047165 | 2.288862 |
| LPE | 0.019736 | 0.045338 |
| PE | 9.930699 | 7.401288 |
| Pathway | Target Protein Name | %CFC |
|---|---|---|
| Lipid metabolism | CASP1 | 27 |
| AcCoA carboxylase | 18 | |
| Glucose metabolism | PP6C | 182 |
| PCK2 | −34 | |
| PyDK2 (PDHK2) | −36 | |
| GSK3a/b | −61 | |
| Inflammation | mTOR (FRAP) | −46 |
| STAT3 | −25 | |
| Chaperone | Hsp60 | −58 |
| Translation | PP6C | 182 |
| Apoptosis | Bad | 94 |
| PTEN | −70 | |
| Cell growth & division | CDK1/2 | −64 |
| Neuro | Tyrosine hydroxylase | −40 |
| Calcium | PKA Ca/b | 163 |
| PTP1D | −58 | |
| CREB | CREB | 122 |
| Jun | JNK | 161 |
| JNK2 | −24 | |
| NF-κB | NF-κB p50 | −49 |
| Orphan | Crystallin αB | 276 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, P.; Cole, L.K.; Chandrakumar, A.; Hauff, K.D.; Ravandi, A.; Dolinsky, V.W.; Hatch, G.M. Phosphokinome Analysis of Barth Syndrome Lymphoblasts Identify Novel Targets in the Pathophysiology of the Disease. Int. J. Mol. Sci. 2018, 19, 2026. https://doi.org/10.3390/ijms19072026
Agarwal P, Cole LK, Chandrakumar A, Hauff KD, Ravandi A, Dolinsky VW, Hatch GM. Phosphokinome Analysis of Barth Syndrome Lymphoblasts Identify Novel Targets in the Pathophysiology of the Disease. International Journal of Molecular Sciences. 2018; 19(7):2026. https://doi.org/10.3390/ijms19072026
Chicago/Turabian StyleAgarwal, Prasoon, Laura K. Cole, Abin Chandrakumar, Kristin D. Hauff, Amir Ravandi, Vernon W. Dolinsky, and Grant M. Hatch. 2018. "Phosphokinome Analysis of Barth Syndrome Lymphoblasts Identify Novel Targets in the Pathophysiology of the Disease" International Journal of Molecular Sciences 19, no. 7: 2026. https://doi.org/10.3390/ijms19072026