Potential for Mitochondrial DNA Sequencing in the Differential Diagnosis of Gynaecological Malignancies

 , ,
, ,
Abstract
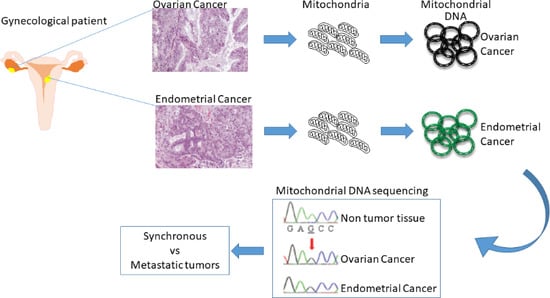
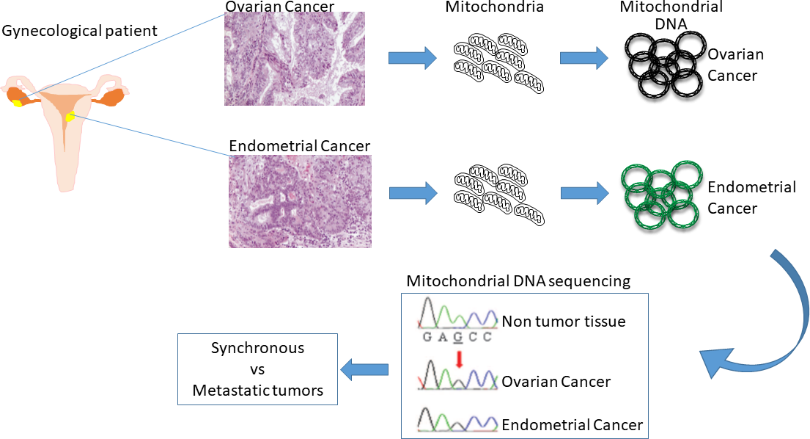
:
1. The Issue of Synchrony in Gynecological Cancers
2. Currently Used Molecular Analyses to Infer Tumor Clonality
3. Mitochondrial DNA Sequencing to Distinguish Simultaneous Versus Metastatic Gynecological Cancers
3.1. Advantages in the Use of Mitochondrial DNA Sequencing
3.2. Endometrial and Ovarian Cancers
3.3. Borderline Ovarian Tumors (BOT) and Peritoneal Implants
3.4. Other Applications of mtDNA Sequencing
4. Discussion and Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Matlock, D.L.; Salem, F.A.; Charles, E.H.; Savage, E.W. Synchronous multiple primary neoplasms of the upper female genital tract. Gynecol. Oncol. 1982, 13, 271–277. [Google Scholar] [CrossRef]
- Ayhan, A.; Yalcin, O.T.; Tuncer, Z.S.; Gurgan, T.; Kucukali, T. Synchronous primary malignancies of the female genital tract. Eur. J. Obstet. Gynecol. Reprod. Biol. 1992, 45, 63–66. [Google Scholar] [CrossRef]
- Singh, N. Synchronous tumours of the female genital tract. Histopathology 2010, 56, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, D.; Ryan, A.; Ayhan, A.; McCluggage, W.G.; Feakins, R.; Santibanez-Koref, M.F.; Mein, C.A.; Gayther, S.A.; Jacobs, I.J. A molecular genetic and statistical approach for the diagnosis of dual-site cancers. J. Natl. Cancer Inst. 2004, 96, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Dizon, D.S.; Birrer, M.J. Making a Difference: Distinguishing Two Primaries from Metastasis in Synchronous Tumors of the Ovary and Uterus. J. Natl. Cancer Inst. 2016, 108, djv442. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Monk, B.J.; Sood, A.K.; Herzog, T.J. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013, 10, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primer 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.-Y.; Lee, Y.-S.; Park, J.-S.; Bae, S.-N.; Lee, J.-M.; Namkoong, S.-E. Clinical analysis of synchronous primary neoplasms of the female reproductive tract. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 136, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Ulbright, T.M.; Roth, L.M. Metastatic and independent cancers of the endometrium and ovary: A clinicopathologic study of 34 cases. Hum. Pathol. 1985, 16, 28–34. [Google Scholar] [CrossRef]
- Kurman, R.J.; Hedrick Ellenson, L.; Ronnett, B.M.; Blaustein, A. (Eds.) Blaustein’s Pathology of the Female Genital Tract: With 1446 Figures and 125 Tables, 6th ed.; Springer: New York, NY, USA; Dodrecht, The Netherlands; Heidelberg, Germany; London, UK, 2011; ISBN 978-1-4419-0488-1. [Google Scholar]
- Scully, R.E.; Young, R.H.; Clement, P.B. Tumors of the Ovary, Maldeveloped Gonads, Fallopian Tube, and Broad Ligament; Atlas of Tumor Pathology; Armed Forces Institute of Pathology: Washington, DC, USA, 1998; ISBN 978-1-881041-43-6. [Google Scholar]
- Seidman, J.D.; Kurman, R.J. Ovarian serous borderline tumors: A critical review of the literature with emphasis on prognostic indicators. Hum. Pathol. 2000, 31, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Schmid, S.; Heinimann, K.; Frick, H.; Herrmann, C.; Cerny, T.; Omlin, A. Multiple primary tumours: Challenges and approaches, a review. ESMO Open 2017, 2, e000172. [Google Scholar] [CrossRef] [PubMed]
- Debska-Szmich, S.; Czernek, U.; Krakowska, M.; Frackowiak, M.; Zieba, A.; Czyzykowski, R.; Kulejewska, D.; Potemski, P. Synchronous primary ovarian and endometrial cancers: A series of cases and a review of literature. Przeglad Menopauzalny Menopause Rev. 2014, 13, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Matias-Guiu, X.; Lagarda, H.; Catasus, L.; Bussaglia, E.; Gallardo, A.; Gras, E.; Prat, J. Clonality analysis in synchronous or metachronous tumors of the female genital tract. Int. J. Gynecol. Pathol. 2002, 21, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Magini, P.; Cormio, A.; Santini, D.; Ceccarelli, C.; Gasparre, G. Mitochondrial DNA genotyping reveals synchronous nature of simultaneously detected endometrial and ovarian cancers. Gynecol. Oncol. 2011, 122, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Girolimetti, G.; Perrone, A.M.; Procaccini, M.; Kurelac, I.; Ceccarelli, C.; De Biase, D.; Caprara, G.; Zamagni, C.; De Iaco, P.; et al. Mitochondrial DNA genotyping efficiently reveals clonality of synchronous endometrial and ovarian cancers. Mod. Pathol. 2014, 27, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girolimetti, G.; De Iaco, P.; Procaccini, M.; Panzacchi, R.; Kurelac, I.; Amato, L.B.; Dondi, G.; Caprara, G.; Ceccarelli, C.; Santini, D.; et al. Mitochondrial DNA sequencing demonstrates clonality of peritoneal implants of borderline ovarian tumors. Mol. Cancer 2017, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Enomoto, T.; Wada, H.; Inoue, M.; Okudaira, Y.; Shroyer, K.R. Application of clonal analysis. Differential diagnosis for synchronous primary ovarian and endometrial cancers and metastatic cancer. Am. J. Clin. Pathol. 1996, 105, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.M.; Forgacs, E.; Warshal, D.P.; Yeh, I.T.; Martin, J.S.; Ashfaq, R.; Muller, C.Y. Loss of heterozygosity and mutational analysis of the PTEN/MMAC1 gene in synchronous endometrial and ovarian carcinomas. Clin. Cancer Res. 1998, 4, 2577–2583. [Google Scholar] [PubMed]
- Fujii, H.; Matsumoto, T.; Yoshida, M.; Furugen, Y.; Takagaki, T.; Iwabuchi, K.; Nakata, Y.; Takagi, Y.; Moriya, T.; Ohtsuji, N.; et al. Genetics of synchronous uterine and ovarian endometrioid carcinoma: Combined analyses of loss of heterozygosity, PTEN mutation, and microsatellite instability. Hum. Pathol. 2002, 33, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Irving, J.A.; Catasus, L.; Gallardo, A.; Bussaglia, E.; Romero, M.; Matias-Guiu, X.; Prat, J. Synchronous endometrioid carcinomas of the uterine corpus and ovary: Alterations in the beta-catenin (CTNNB1) pathway are associated with independent primary tumors and favorable prognosis. Hum. Pathol. 2005, 36, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Nakamura, K.; Nomura, H.; Banno, K.; Irie, H.; Adachi, M.; Iida, M.; Umene, K.; Nogami, Y.; Masuda, K.; et al. Clinicopathologic analysis with immunohistochemistry for DNA mismatch repair protein expression in synchronous primary endometrial and ovarian cancers. Int. J. Gynecol. Cancer 2015, 25, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.L.; Mutter, G.L. Molecular and pathologic aspects of endometrial carcinogenesis. J. Clin. Oncol. 2006, 24, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Kaneki, E.; Oda, Y.; Ohishi, Y.; Tamiya, S.; Oda, S.; Hirakawa, T.; Nakano, H.; Tsuneyoshi, M. Frequent microsatellite instability in synchronous ovarian and endometrial adenocarcinoma and its usefulness for differential diagnosis. Hum. Pathol. 2004, 35, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Shenson, D.L.; Gallion, H.H.; Powell, D.E.; Pieretti, M. Loss of heterozygosity and genomic instability in synchronous endometrioid tumors of the ovary and endometrium. Cancer 1995, 76, 650–657. [Google Scholar] [CrossRef] [Green Version]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, C.; Kirk, J.; Barnetson, R.; Evans, J.; Schnitzler, M.; Quinn, M.; Hacker, N.; Crandon, A.; Harnett, P. Incidence of microsatellite instability in synchronous tumors of the ovary and endometrium. Clin. Cancer Res. 2003, 9, 1387–1392. [Google Scholar] [PubMed]
- Singer, G.; Kallinowski, T.; Hartmann, A.; Dietmaier, W.; Wild, P.J.; Schraml, P.; Sauter, G.; Mihatsch, M.J.; Moch, H. Different types of microsatellite instability in ovarian carcinoma. Int. J. Cancer 2004, 112, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plisiecka-Hałasa, J.; Dansonka-Mieszkowska, A.; Kraszewska, E.; Dańska-Bidzińska, A.; Kupryjańczyk, J. Loss of heterozygosity, microsatellite instability and TP53 gene status in ovarian carcinomas. Anticancer Res. 2008, 28, 989–996. [Google Scholar] [PubMed]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.P.; Markowitz, S.; Willson, J.K.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halperin, R.; Zehavi, S.; Hadas, E.; Habler, L.; Bukovsky, I.; Schneider, D. Simultaneous carcinoma of the endometrium and ovary vs endometrial carcinoma with ovarian metastases: A clinical and immunohistochemical determination. Int. J. Gynecol. Cancer 2003, 13, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, M. Wnt signalling and the mechanistic basis of tumour development. J. Pathol. 2005, 205, 130–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.-H.; Albarracin, C.; Luthra, R.; Wang, L.; Zheng, W.; Malpica, A.; Deavers, M.T.; Silva, E.G.; Liu, J. Discordant genetic changes in ovarian and endometrial endometrioid carcinomas: A potential pitfall in molecular diagnosis. Int. J. Gynecol. Cancer 2006, 16, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Goodheart, M.J.; Ritchie, J.M.; Rose, S.L.; Fruehauf, J.P.; De Young, B.R.; Buller, R.E. The relationship of molecular markers of p53 function and angiogenesis to prognosis of stage I epithelial ovarian cancer. Clin. Cancer Res. 2005, 11, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Dinjens, W.N.M.; van der Burg, M.E.L.; Chadha, S.; Sleddens, H.F.B.M.; Burger, C.W.; Ewing, P.C. Clinical importance of molecular determinations in gynecologic patients with multiple tumors. Cancer 2003, 97, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, J. Clonality analysis in synchronous tumors of the female genital tract. Hum. Pathol. 2002, 33, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, A.M.; Ng, C.K.Y.; De Filippo, M.R.; Piscuoglio, S.; Macedo, G.S.; Gatius, S.; Perez Mies, B.; Soslow, R.A.; Lim, R.S.; Viale, A.; et al. Massively Parallel Sequencing-Based Clonality Analysis of Synchronous Endometrioid Endometrial and Ovarian Carcinomas. J. Natl. Cancer Inst. 2016, 108, djv427. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Wang, Y.K.; Maassen, M.; Horlings, H.M.; Bashashati, A.; Senz, J.; Mackenzie, R.; Grewal, D.S.; Li-Chang, H.; Karnezis, A.N.; et al. Synchronous Endometrial and Ovarian Carcinomas: Evidence of Clonality. J. Natl. Cancer Inst. 2016, 108, djv428. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, N.; Lang, F.M.; Noske, A.; Samartzis, E.P.; Schmidt, A.-M.; Bellini, E.; Fink, D.; Moch, H.; Rechsteiner, M.; Dedes, K.J.; et al. Tracking the origin of simultaneous endometrial and ovarian cancer by next-generation sequencing—A case report. BMC Cancer 2017, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.J.; Hoivik, E.A.; Halle, M.K.; Taylor-Weiner, A.; Cherniack, A.D.; Berg, A.; Holst, F.; Zack, T.I.; Werner, H.M.J.; Staby, K.M.; et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 2016, 48, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.-C.; Veras, E.; Lin, J.; Gerry, E.; Bahadirli-Talbott, A.; Baras, A.; Ayhan, A.; Shih, I.-M.; Wang, T.-L. Elucidating the pathogenesis of synchronous and metachronous tumors in a woman with endometrioid carcinomas using a whole-exome sequencing approach. Cold Spring Harb. Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Jing, Y.; Cai, M.-C.; Ma, P.; Zhang, Y.; Xu, C.; Zhang, M.; Di, W.; Zhuang, G. Clonality, Heterogeneity, and Evolution of Synchronous Bilateral Ovarian Cancer. Cancer Res. 2017, 77, 6551–6561. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Gamallo, C.; Pérez-Gallego, L.; de Mora, J.C.; Suárez, A.; Palacios, J. β-Catenin expression pattern, β-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagn. Mol. Pathol. 2001, 10, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, B.H.; Ailawadi, M.; Colitti, C.; Muto, M.G.; Deavers, M.; Silva, E.G.; Berkowitz, R.S.; Mok, S.C.; Gershenson, D.M. Second primary or recurrence? Comparative patterns of p53 and K-ras mutations suggest that serous borderline ovarian tumors and subsequent serous carcinomas are unrelated tumors. Cancer Res. 2001, 61, 7264–7267. [Google Scholar] [PubMed]
- Sieben, N.L.; Kolkman-Uljee, S.M.; Flanagan, A.M.; le Cessie, S.; Cleton-Jansen, A.M.; Cornelisse, C.J.; Fleuren, G.J. Molecular genetic evidence for monoclonal origin of bilateral ovarian serous borderline tumors. Am. J. Pathol. 2003, 162, 1095–1101, Erratum in: Am. J. Pathol. 2006, 168, 714. [Google Scholar] [CrossRef]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clima, R.; Preste, R.; Calabrese, C.; Diroma, M.A.; Santorsola, M.; Scioscia, G.; Simone, D.; Shen, L.; Gasparre, G.; Attimonelli, M. HmtDB 2016: Data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res. 2017, 45, D698–D706. [Google Scholar] [CrossRef] [PubMed]
- Santorsola, M.; Calabrese, C.; Girolimetti, G.; Diroma, M.A.; Gasparre, G.; Attimonelli, M. A multi-parametric workflow for the prioritization of mitochondrial DNA variants of clinical interest. Hum. Genet. 2016, 135, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Shen, L.; Gonzalez, M.; Leipzig, J.; Lott, M.T.; Stassen, A.P.M.; Diroma, M.A.; Navarro-Gomez, D.; Yeske, P.; Bai, R.; et al. Mitochondrial Disease Sequence Data Resource (MSeqDR): A global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol. Genet. Metab. 2015, 114, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, S.; Shoshan, M. (Eds.) Tumor Cell Metabolism: Pathways, Regulation and Biology; Springer: Wien, Austria, 2015; ISBN 978-3-7091-1824-5. [Google Scholar]
- Iommarini, L.; Calvaruso, M.A.; Kurelac, I.; Gasparre, G.; Porcelli, A.M. Complex I impairment in mitochondrial diseases and cancer: Parallel roads leading to different outcomes. Int. J. Biochem. Cell Biol. 2013, 45, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.-D.; Sun, Y.-B.; Li, E.-M.; Xu, L.-Y.; Zhang, Y.-P.; Yao, Y.-G.; Kong, Q.-P. Deciphering the signature of selective constraints on cancerous mitochondrial genome. Mol. Biol. Evol. 2012, 29, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Foschini, M.P.; Morandi, L.; Marchetti, C.; Cocchi, R.; Eusebi, L.H.; Farnedi, A.; Badiali, G.; Gissi, D.B.; Pennesi, M.G.; Montebugnoli, L. Cancerization of cutaneous flap reconstruction for oral squamous cell carcinoma: report of three cases studied with the mtDNA D-loop sequence analysis. Histopathology 2011, 58, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Fellous, T.G.; McDonald, S.A.C.; Burkert, J.; Humphries, A.; Islam, S.; De-Alwis, N.M.W.; Gutierrez-Gonzalez, L.; Tadrous, P.J.; Elia, G.; Kocher, H.M.; et al. A methodological approach to tracing cell lineage in human epithelial tissues. Stem Cells 2009, 27, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Hibar, D.P.; Stein, J.L.; Renteria, M.E.; Arias-Vasquez, A.; Desrivieres, S.; Jahanshad, N.; Toro, R.; Wittfeld, K.; Abramovic, L.; Andersson, M.; et al. Common genetic variants influence human subcortical brain structures. Nature 2015, 520, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bese, T.; Sal, V.; Kahramanoglu, I.; Tokgozoglu, N.; Demirkiran, F.; Turan, H.; Ilvan, S.; Arvas, M. Synchronous Primary Cancers of the Endometrium and Ovary With the Same Histopathologic Type Versus Endometrial Cancer With Ovarian Metastasis: A Single Institution Review of 72 Cases. Int. J. Gynecol. Cancer 2016, 26, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhao, Q.; Lv, X. Characteristics and prognosis of coexisting adnexa malignancy with endometrial cancer: A single institution review of 51 cases. Arch. Gynecol. Obstet. 2011, 283, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Jin, H.; Lu, Y.; Lu, X. Clinicopathological characteristics of patients with synchronous primary endometrial and ovarian cancers: A review of 43 cases. Oncol. Lett. 2013, 5, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Seong, S.J.; Bae, D.-S.; Kim, J.-H.; Suh, D.H.; Lee, K.-H.; Park, S.-Y.; Lee, T.S. Prognostic factors in women with synchronous endometrial and ovarian cancers. Int. J. Gynecol. Cancer 2014, 24, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Sozen, H.; Vatansever, D.; Iyibozkurt, A.C.; Topuz, S.; Ozsurmeli, M.; Salihoglu, Y.; Guzelbey, B.; Berkman, S. Clinicopathologic and survival analyses of synchronous primary endometrial and epithelial ovarian cancers. J. Obstet. Gynaecol. Res. 2015, 41, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Tejerizo-Garcia, A.; Jimenez-Lopez, J.S.; Munoz-Gonzalez, J.L.; Bartolome-Sotillos, S.; Marqueta-Marques, L.; Lopez-Gonzalez, G.; Gomez, J.F.P.-R. Overall survival and disease-free survival in endometrial cancer: Prognostic factors in 276 patients. OncoTargets Ther. 2013, 9, 1305–1313. [Google Scholar] [CrossRef]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; Gonzalez-Martin, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO consensus conference on endometrial cancer: Diagnosis, treatment and follow-up. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2015, 117, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A. First-line chemotherapy in epithelial ovarian cancer. Clin. Obstet. Gynecol. 2012, 55, 96–113. [Google Scholar] [CrossRef] [PubMed]
- Heitz, F.; Amant, F.; Fotopoulou, C.; Battista, M.J.; Wimberger, P.; Traut, A.; Fisseler-Eckhoff, A.; Harter, P.; Vandenput, I.; Sehouli, J.; et al. Synchronous ovarian and endometrial cancer—An international multicenter case-control study. Int. J. Gynecol. Cancer 2014, 24, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, L.E.; Rambau, P.F.; Koziak, J.M.; Steed, H.; Köbel, M. Synchronous endometrial and ovarian carcinomas: Predictors of risk and associations with survival and tumor expression profiles. Cancer Causes Control. 2017, 28, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Tropé, C.G.; Kaern, J.; Davidson, B. Borderline ovarian tumours. Best Pract. Res. Clin. Obstet. Gynaecol. 2012, 26, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Menczer, J.; Chetrit, A.; Sadetzki, S. The effect of hysterectomy on survival of patients with borderline ovarian tumors. Gynecol. Oncol. 2012, 125, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, N.; Shanbhogue, A.K.P.; Vikram, R.; Nagar, A.; Jagirdar, J.; Prasad, S.R. Current update on borderline ovarian neoplasms. AJR Am. J. Roentgenol. 2010, 194, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.G.; Gershenson, D.M.; Malpica, A.; Deavers, M. The recurrence and the overall survival rates of ovarian serous borderline neoplasms with noninvasive implants is time dependent. Am. J. Surg. Pathol. 2006, 30, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Sieben, N.L.; Roemen, G.M.J.M.; Oosting, J.; Fleuren, G.J.; van Engeland, M.; Prat, J. Clonal analysis favours a monoclonal origin for serous borderline tumours with peritoneal implants. J. Pathol. 2006, 210, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Bell, D.A.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Evidence for the multifocal origin of bilateral and advanced human serous borderline ovarian tumors. Cancer Res. 1998, 58, 2328–2330. [Google Scholar] [PubMed]
- Mok, C.H.; Tsao, S.W.; Knapp, R.C.; Fishbaugh, P.M.; Lau, C.C. Unifocal origin of advanced human epithelial ovarian cancers. Cancer Res. 1992, 52, 5119–5122. [Google Scholar] [PubMed]
- Tsao, S.W.; Mok, C.H.; Knapp, R.C.; Oike, K.; Muto, M.G.; Welch, W.R.; Goodman, H.M.; Sheets, E.E.; Berkowitz, R.S.; Lau, C.C. Molecular genetic evidence of a unifocal origin for human serous ovarian carcinomas. Gynecol. Oncol. 1993, 48, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Abeln, E.C.; Kuipers-Dijkshoorn, N.J.; Berns, E.M.; Henzen-Logmans, S.C.; Fleuren, G.J.; Cornelisse, C.J. Molecular genetic evidence for unifocal origin of advanced epithelial ovarian cancer and for minor clonal divergence. Br. J. Cancer 1995, 72, 1330–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchion, D.C.; Xiong, Y.; Chon, H.S.; Al Sawah, E.; Bou Zgheib, N.; Ramirez, I.J.; Abbasi, F.; Stickles, X.B.; Judson, P.L.; Hakam, A.; et al. Gene expression data reveal common pathways that characterize the unifocal nature of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 576.e1–576.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, I.-M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Ozols, R.F.; Bookman, M.A.; Connolly, D.C.; Daly, M.B.; Godwin, A.K.; Schilder, R.J.; Xu, X.; Hamilton, T.C. Focus on epithelial ovarian cancer. Cancer Cell 2004, 5, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Naora, H.; Montell, D.J. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.; Bannatyne, P.M.; Solomon, H.J.; Stoddard, L.D.; Tattersall, M.H. Multifocal tumorigenesis in the upper female genital tract—Implications for staging and management. Int. J. Gynecol. Pathol. 1985, 4, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, J.D.; Solomon, D.; Sullivant, H. Multifocal disease in the upper genital canal. Obstet. Gynecol. 1985, 65, 695–698. [Google Scholar] [PubMed]
- Park, T.W.; Felix, J.C.; Wright, T.C.J. X chromosome inactivation and microsatellite instability in early and advanced bilateral ovarian carcinomas. Cancer Res. 1995, 55, 4793–4796. [Google Scholar] [PubMed]
- Pejovic, T.; Heim, S.; Mandahl, N.; Elmfors, B.; Furgyik, S.; Floderus, U.M.; Helm, G.; Willen, H.; Mitelman, F. Bilateral ovarian carcinoma: Cytogenetic evidence of unicentric origin. Int. J. Cancer 1991, 47, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Cerny, T.; Savidan, A.; Vounatsou, P.; Konzelmann, I.; Bouchardy, C.; Frick, H.; Ess, S. Cancer survivors in Switzerland: A rapidly growing population to care for. BMC Cancer 2013, 13, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amer, M.H. Multiple neoplasms, single primaries, and patient survival. Cancer Manag. Res. 2014, 6, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.M.; Girolimetti, G.; Cima, S.; Kurelac, I.; Livi, A.; Caprara, G.; Santini, D.; Castellucci, P.; Morganti, A.G.; Gasparre, G.; et al. Pathological and molecular diagnosis of bilateral inguinal lymph nodes metastases from low-grade endometrial adenocarcinoma: A case report with review of the literature. BMC Cancer 2018, 18, 7. [Google Scholar] [CrossRef] [PubMed]
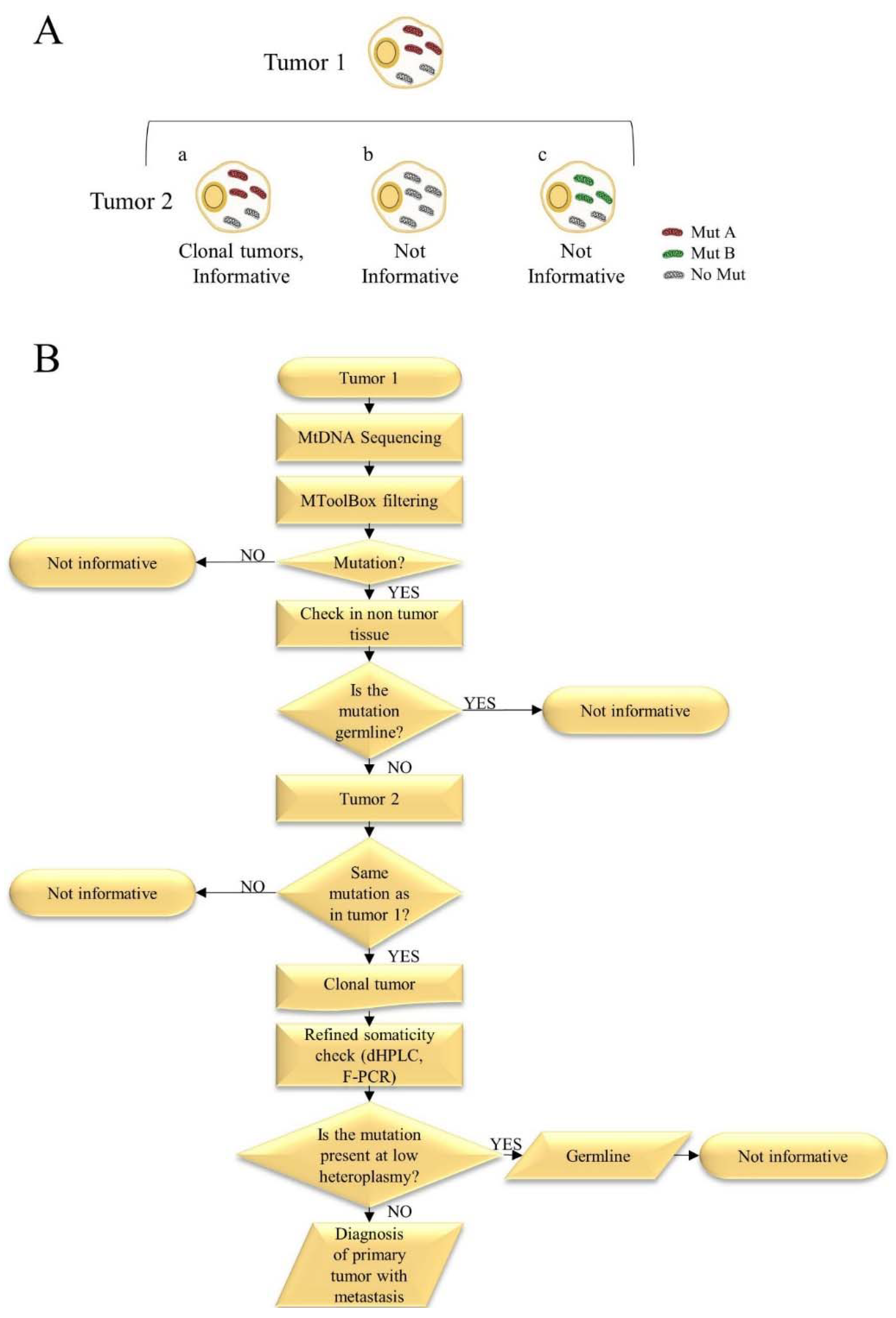
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Gutta, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. MToolBox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Wu, R.C.; Jung, S.M.; Lee, Y.S.; Chen, S.J.; Lu, Y.L.; Tsai, C.L.; Lin, C.Y.; Tang, Y.H.; Chen, M.Y.; et al. Implication of genomic characterization in synchronous endometrial and ovarian cancers of endometrioid histology. Gynecol. Oncol. 2016, 143, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Kurelac, I.; Lang, M.; Zuntini, R.; Calabrese, C.; Simone, D.; Vicario, S.; Santamaria, M.; Attimonelli, M.; Romeo, G.; Gasparre, G. Searching for a needle in the haystack: Comparing six methods to evaluate heteroplasmy in difficult sequence context. Biotechnol. Adv. 2012, 30, 363–371. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Technique | Efficiency | Type of Information |
|---|---|---|
| MSI | 18% [17] 76% [25] 17–32% (EC) [30] 3–17% (OC) [30] | IT/CT |
| B-Catenin IHC | 38% [47] 45.5% [17] | IT |
| CTNNB1 mutation screening | 44% [22] 25–38% [24] | IT/CT |
| Tumor suppressor mutation screening | 30–50% [21] 88% [48] | IT/CT |
| X-chromosome inactivation | 27% [25] | IT |
| LOH pattern | 53% [5] 41% [49] | IT/CT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrone, A.M.; Girolimetti, G.; Procaccini, M.; Marchio, L.; Livi, A.; Borghese, G.; Porcelli, A.M.; De Iaco, P.; Gasparre, G. Potential for Mitochondrial DNA Sequencing in the Differential Diagnosis of Gynaecological Malignancies. Int. J. Mol. Sci. 2018, 19, 2048. https://doi.org/10.3390/ijms19072048
Perrone AM, Girolimetti G, Procaccini M, Marchio L, Livi A, Borghese G, Porcelli AM, De Iaco P, Gasparre G. Potential for Mitochondrial DNA Sequencing in the Differential Diagnosis of Gynaecological Malignancies. International Journal of Molecular Sciences. 2018; 19(7):2048. https://doi.org/10.3390/ijms19072048
Chicago/Turabian StylePerrone, Anna Myriam, Giulia Girolimetti, Martina Procaccini, Lorena Marchio, Alessandra Livi, Giulia Borghese, Anna Maria Porcelli, Pierandrea De Iaco, and Giuseppe Gasparre. 2018. "Potential for Mitochondrial DNA Sequencing in the Differential Diagnosis of Gynaecological Malignancies" International Journal of Molecular Sciences 19, no. 7: 2048. https://doi.org/10.3390/ijms19072048