Genome-Wide Characterization and Expression Analyses of Pleurotus ostreatus MYB Transcription Factors during Developmental Stages and under Heat Stress Based on de novo Sequenced Genome


 and
and
Abstract
:1. Introduction
2. Results
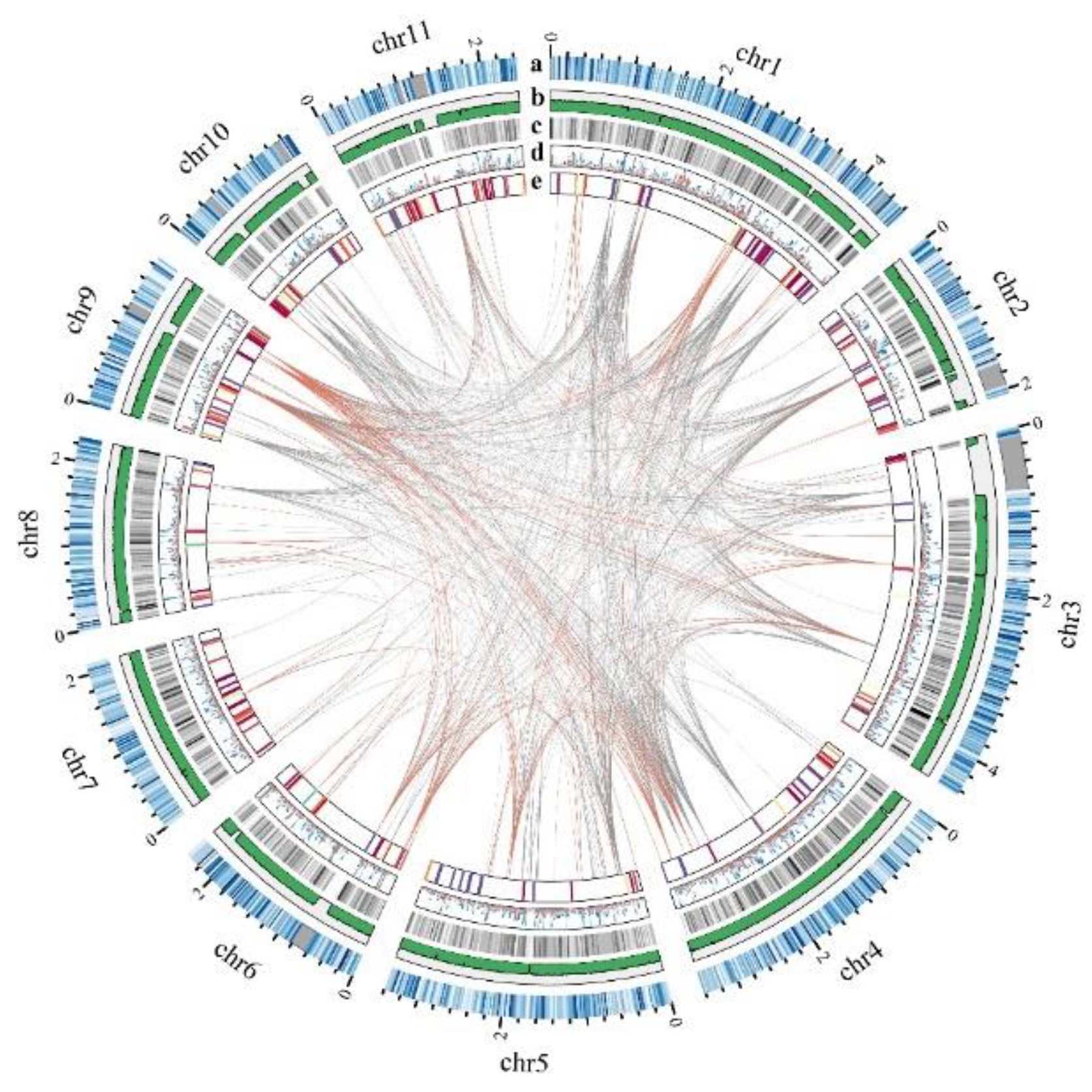
2.1. Genome Sequencing, Assembly, and Annotation
2.2. Identification and Classification of MYB Genes in P. ostreatus
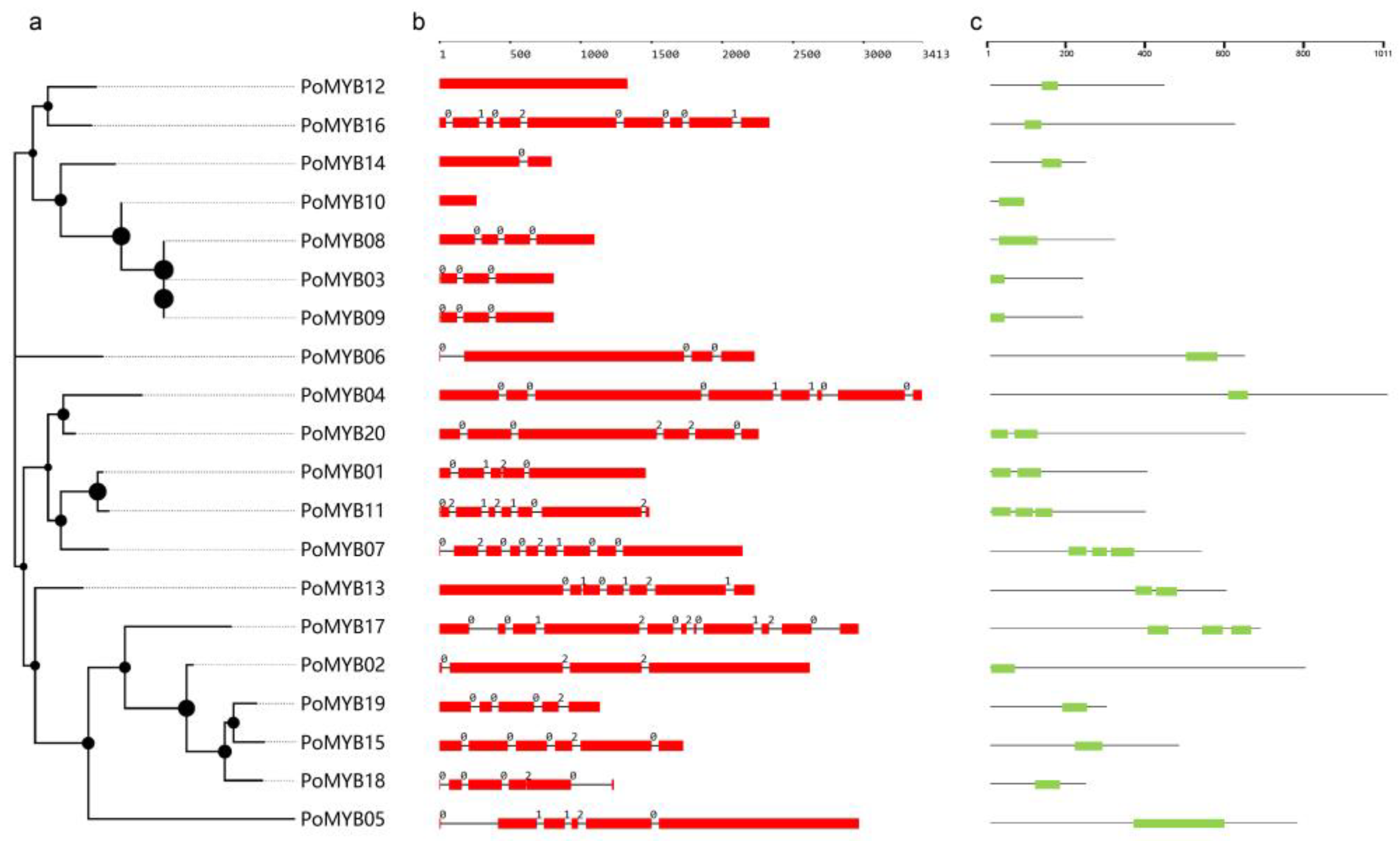
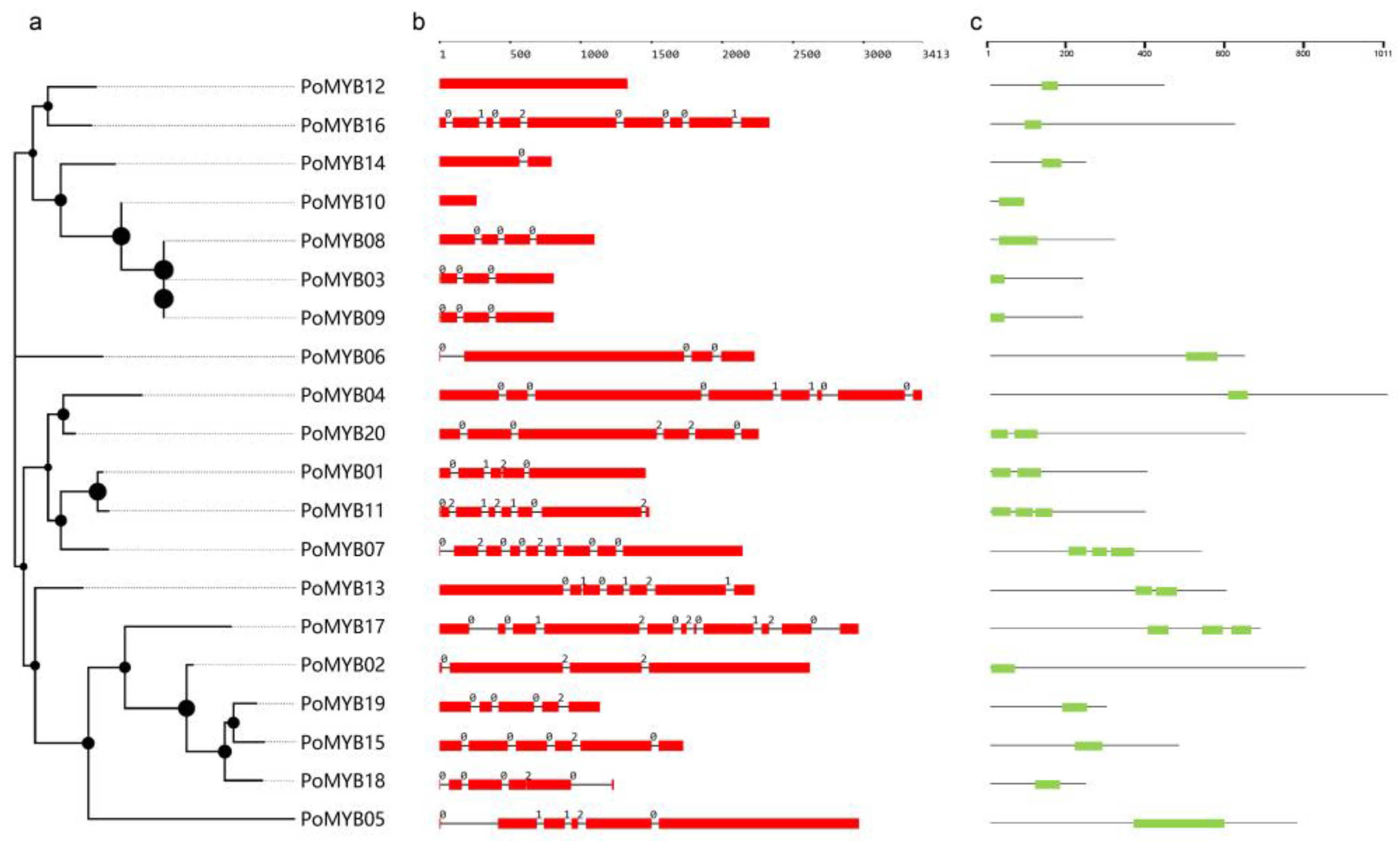
2.3. Sequence Analysis, Gene Structure, and Phylogenetic Analysis of PoMYBs
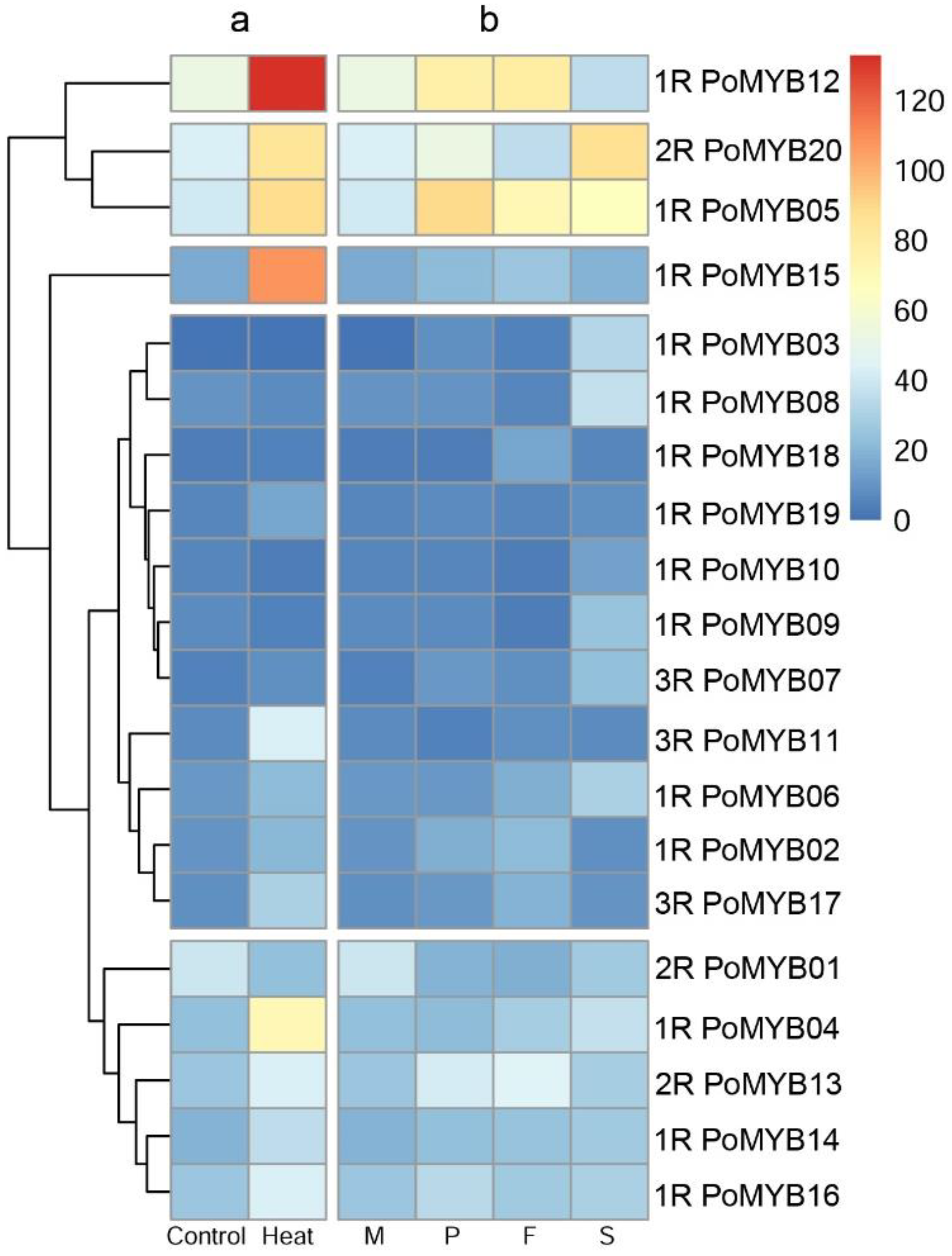
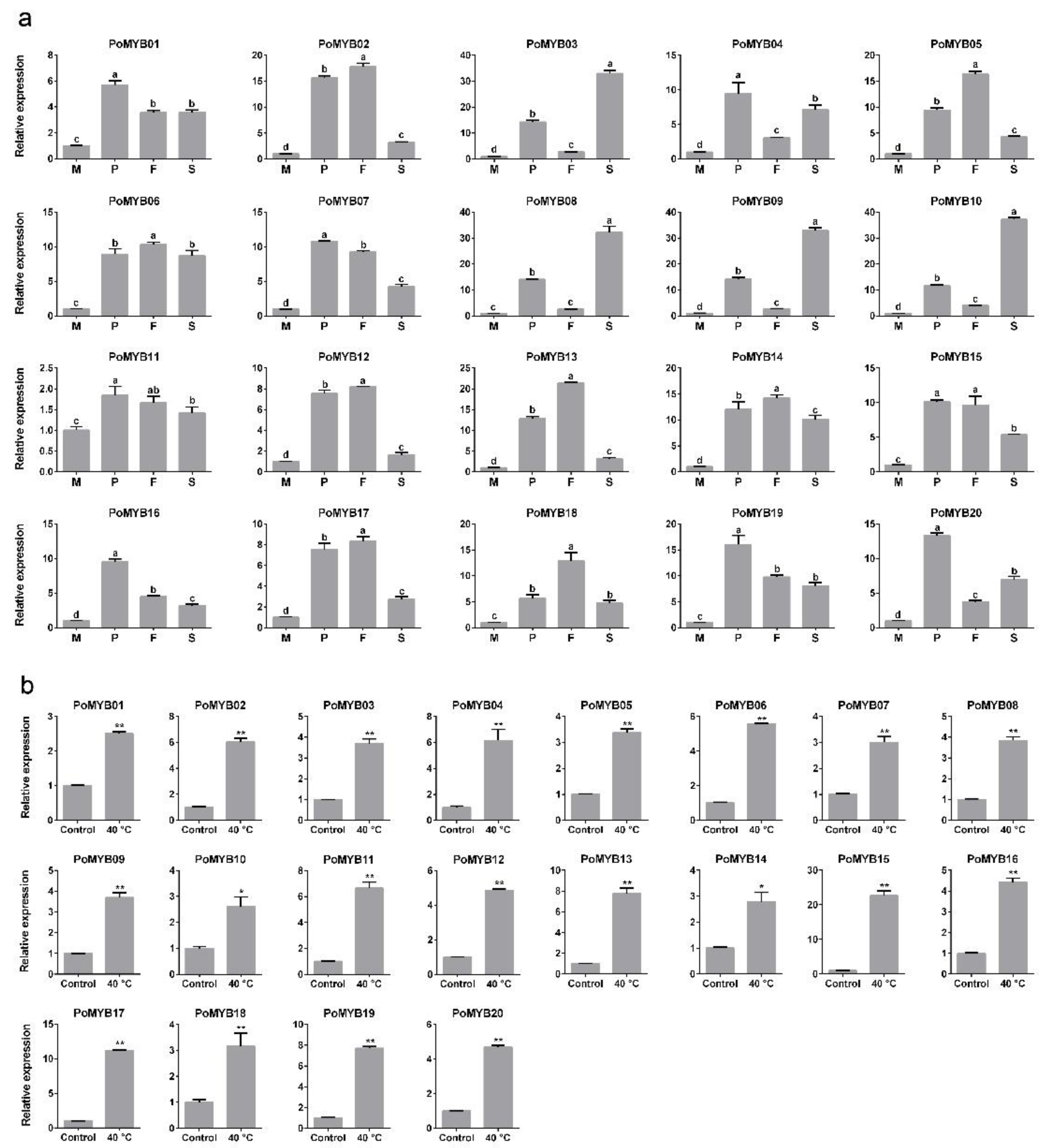
2.4. Expression of PoMYBs during Different Developmental Stages and under Heat Stress of P. ostreatus
3. Discussion
4. Materials and Methods
4.1. Strains and Culture Conditions
4.2. Genome Sequencing, Assembly, and Annotation
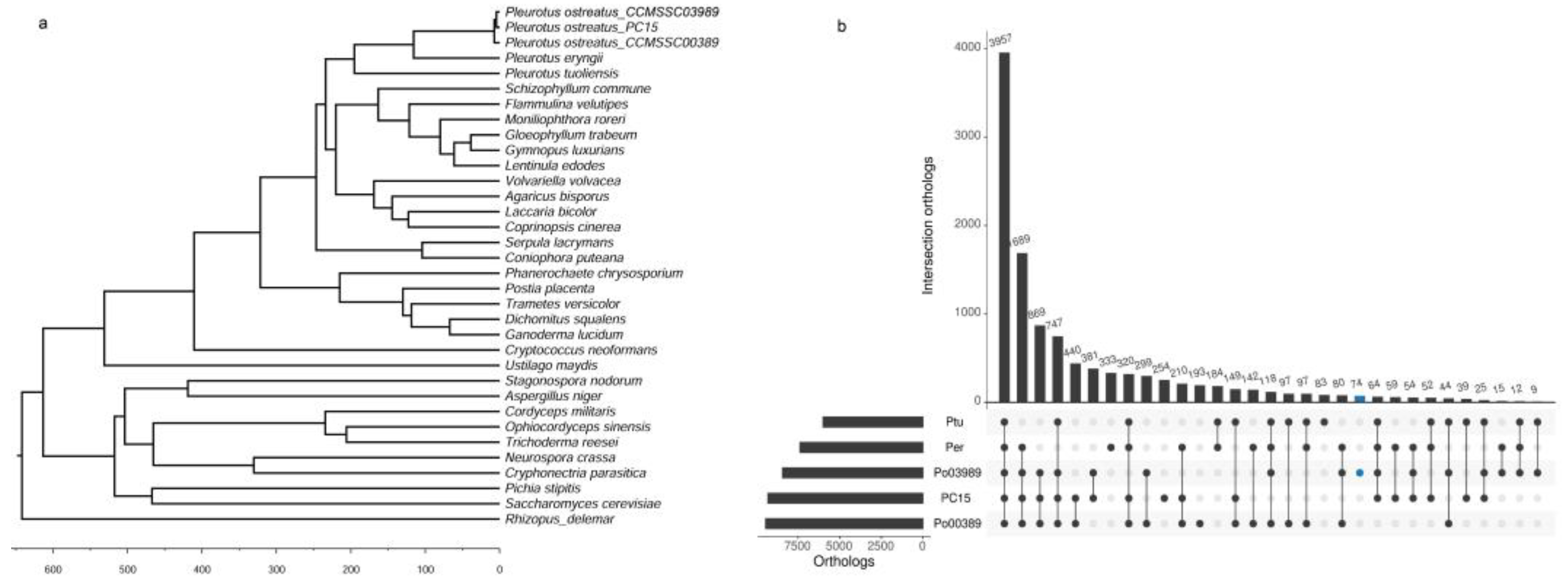
4.3. Phylogenetic Analysis and Molecular Clock Estimation
4.4. Identification, Cloning, and Classification of the MYB Gene Family in P. ostreatus
4.5. Sequence Analysis, Gene Structure, and Phylogenetic Analysis of MYB Genes
4.6. RNA-Sequencing and Analysis
4.7. Expression Analysis by qPCR
4.8. Data Availability and Accession Numbers
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-generation sequencing |
| BUSCO | Benchmarking universal single-copy orthologs |
| KAT | K-mer analysis toolkit |
| ML | Maximum likelihood |
| MRCA | Most recent common ancestor |
| Mya | Million years ago |
| RNA-Seq | RNA-sequencing |
| FPKM | Fragments per kilobase of exon per million mapped reads |
| qPCR | Quantitative PCR |
References
- Atri, N.; Sharma, S.K.; Joshi, R.; Gulati, A.; Gulati, A. Nutritional and neutraceutical composition of five wild culinary-medicinal species of genus Pleurotus (higher Basidiomycetes) from northwest India. Int. J. Med. Mushrooms 2013, 15, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C. Cultivation of Pleurotus ostreatus and other edible mushrooms. Appl. Microbiol. Biotechnol. 2010, 85, 1321–1337. [Google Scholar] [CrossRef] [PubMed]
- Eger, G.; Eden, G.; Wissig, E. Pleurotus ostreatus—Breeding potential of a new cultivated mushroom. Theor. Appl. Genet. 1976, 47, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Zhou, G.Y.; Ma, J.X.; Jiang, W.K.; Jin, L.G.; Zhang, Z.H.; Guo, Y.; Zhang, J.B.; Sui, Y.; Zheng, L.T. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.; Salamov, A.A.; Brown, D.W.; Nagy, L.G.; Floudas, D.; Held, B.W.; Levasseur, A.; Lombard, V.; Morin, E.; Otillar, R.; et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 9923–9928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.B.; Zhao, M.R.; Hsiang, T.; Feng, X.X.; Zhang, J.X.; Huang, C.Y. Identification and characterization of small noncoding RNAs in genome sequences of the edible fungus Pleurotus ostreatus. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Castanera, R.; López-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Pérez, G.; Pisabarro, A.G.; Grigoriev, I.V.; et al. Transposable elements versus the fungal genome: Impact on whole-genome architecture and transcriptional profiles. PLoS Genet. 2016, 12, e1006108. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, M.; Castanera, R.; Lavín, J.L.; Grigoriev, I.V.; Oguiza, J.A.; Ramírez, L.; Pisabarro, A.G. Comparative and transcriptional analysis of the predicted secretome in the lignocellulose-degrading basidiomycete fungus Pleurotus ostreatus. Environ. Microbiol. 2016, 18, 4710–4726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.W.; Lan, Y.F.; Tang, L.N.; Li, X.M. Comparative study on comprehensive agronomic characters of five oyster strains. Shandong Agr. Sci. 2014, 46, 58–59. (In Chinese) [Google Scholar] [CrossRef]
- Im, C.H.; Kim, K.H.; Je, H.J.; Ali, A.; Kim, M.K.; Joung, W.K.; Lee, S.D.; Shin, H.; Ryu, J.S. Multiplex simple sequence repeat (SSR) markers discriminating Pleurotus eryngii cultivar. Korean J. Mycol. 2014, 42, 159–164. [Google Scholar] [CrossRef]
- Gao, W.; Qu, J.; Zhang, J.; Sonnenberg, A.; Chen, Q.; Zhang, Y.; Huang, C. A genetic linkage map of Pleurotus tuoliensis integrated with physical mapping of the de novo sequenced genome and the mating type loci. BMC Genom. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.P.; Gong, M.; Zheng, H.J.; Chen, M.J.; Zhang, L.; Wang, H.; Jiang, J.P.; Wu, L.; Zhu, Y.Q.; Zhu, G.; et al. Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS ONE 2013, 8, e58294. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef] [PubMed]
- Kurata, A.; Fukuta, Y.; Mori, M.; Kishimoto, N.; Shirasaka, N. Draft genome sequence of the Basidiomycetous fungus Flammulina velutipes TR19. Genome Announc. 2016, 4, e00505-e16. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Gong, Y.H.; Cai, Y.L.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.Y.; Liu, Y.; Lei, X.Y.; Wang, G.Z.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.J.; Wang, H.; Wang, J.F.; Bao, D.P. Microsatellites in the genome of the edible mushroom, Volvariella volvacea. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Joh, J.H.; Lee, S.H.; Lee, J.S.; Kim, K.H.; Jeong, S.J.; Youn, W.H.; Kim, N.K.; Son, E.S.; Cho, Y.S.; Yoo, Y.B.; et al. Isolation of genes expressed during the developmental stages of the oyster mushroom, Pleurotus ostreatus, using expressed sequence tags. FEMS Microbiol. Lett. 2007, 276, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Au, C.H.; Wilke, S.K.; Stajich, J.E.; Zolan, M.E.; Pukkila, P.J.; Kwan, H.S. 5′-Serial Analysis of Gene Expression studies reveal a transcriptomic switch during fruiting body development in Coprinopsis cinerea. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.J.; Zhang, L.; Wang, R.Q.; Xie, B.; Li, X.; Chen, R.L.; Guo, L.X.; Xie, B.G. The sequence characteristics and expression models reveal superoxide dismutase involved in cold response and fruiting body development in Volvariella volvacea. Int. J. Mol. Sci. 2016, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.E.; Horton, J.S. Mushrooms by magic: Making connections between signal transduction and fruiting body development in the basidiomycete fungus Schizophyllum commune. FEMS Microbiol. Lett. 2006, 262, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Wu, X.L.; Zhang, J.X.; Wang, H.X.; Huang, C.Y. Gene cloning, expression, and characterization of trehalose-6-phosphate synthase from Pleurotus ostreatus. J. Basic Microb. 2017, 57, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Shang, X.D.; Liu, J.Y.; Tan, Q. Changes in trehalose content, enzyme activity and gene expression related to trehalose metabolism in Flammulina velutipes under heat shock. Microbiology 2016, 162, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Wu, X.L.; Gao, W.; Zhao, M.R.; Zhang, J.X.; Huang, C.Y. Differential expression patterns of Pleurotus ostreatus catalase genes during developmental stages and under heat stress. Genes 2017, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, S.; Chun, J.; Lee, H.; Lee, J. Influence of heat treatment on the antioxidant activities and polyphenolic compounds of Shiitake (Lentinus edodes) mushroom. Food Chem. 2006, 99, 381–387. [Google Scholar] [CrossRef]
- Kong, W.W.; Huang, C.Y.; Chen, Q.; Zou, Y.J.; Zhang, J.X. Nitric oxide alleviates heat stress-induced oxidative damage in Pleurotus eryngii var. tuoliensis. Fungal Genet. Biol. 2012, 49, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.W.; Huang, C.Y.; Chen, Q.; Zou, Y.J.; Zhao, M.R.; Zhang, J.X. Nitric oxide is involved in the regulation of trehalose accumulation under heat stress in Pleurotus eryngii var. tuoliensis. Biotechnol. Lett. 2012, 34, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Chen, Q.; Wu, X.L.; Zhang, J.X.; Huang, C.Y. Heat stress induces apoptotic-like cell death in two Pleurotus species. Curr. Microbiol. 2014, 69, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, S.Y.; Kim, K.S.; Rho, H.S.; Chi, M.H.; Choi, J.; Park, J.; Kong, S.; Park, J.; Goh, J. Homeobox transcription factors are required for conidiation and appressorium development in the rice blast fungus Magnaporthe oryzae. PLoS Genet. 2009, 5, e1000757. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Han, K.Y.; Yu, J.H.; Chae, K.S.; Jahng, K.Y.; Han, D.M. The nsdD gene encodes a putative GATA-type transcription factor necessary for sexual development of Aspergillus nidulans. Mol. Microbiol. 2001, 41, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Vihervaara, A.; Sistonen, L. HSF1 at a glance. J. Cell Sci. 2014, 127, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Wu, Y.X.; Ho, H.; Zhang, H.; He, P.F.; He, Y.Q. BZcon1, a SANT/MYB-type gene involved in the conidiation of Cochliobolus carbonum. G3 Genes Genom. Genet. 2014, 4, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Son, H.; Choi, G.J.; Kim, J.C.; Lee, Y.-W. MYT3, a MYB-like transcription factor, affects fungal development and pathogenicity of Fusarium graminearum. PLoS ONE 2014, 9, e94359. [Google Scholar] [CrossRef] [PubMed]
- Han, X.Y.; Yin, Q.G.; Liu, J.Y.; Jiang, W.B.; Di, S.K.; Pang, Y.Z. GmMYB58 and GmMYB205 are seed-specific activators for isoflavonoid biosynthesis in Glycine max. Plant Cell Rep. 2017, 36, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.X.; Yan, B.Y.; Hu, J.; Yang, C.P.; Li, Y.J.; Liu, J.P.; Liao, W.B. Transcriptome profiling of rubber tree (Hevea brasiliensis) discovers candidate regulators of the cold stress response. Genes Genom. 2018, 41. [Google Scholar] [CrossRef]
- Roy, S. Function of MYB domain transcription factors in abiotic stress and epigenetic control of stress response in plant genome. Plant Signal. Behav. 2016, 11, e1117723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, J.; Tao, K.; Ye, W.W.; Li, A.N.; Liu, X.Y.; Kong, L.; Dong, S.M.; Zheng, X.B.; Wang, Y.C. A MYB transcription factor of Phytophthora sojae, regulated by MAP kinase PsSAK1, is required for zoospore development. PLoS ONE 2012, 7, e40246. [Google Scholar] [CrossRef] [PubMed]
- Arratia-Quijada, J.; Sánchez, O.; Scazzocchio, C.; Aguirre, J. FlbD, a MYB transcription factor of Aspergillus nidulans, is uniquely involved in both asexual and sexual differentiation. Eukaryot. Cell 2012, 11, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A.; Etxebeste, O.; Herrero-Garcia, E.; Ugalde, U.; Espeso, E.A. The concerted action of bZip and cMYB transcription factors FlbB and FlbD induces brlA expression and asexual development in Aspergillus nidulans. Mol. Microbiol. 2010, 75, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Mateos, L.; Jiménez, A.; Revuelta, J.L.; Santos, M.A. Purine biosynthesis, riboflavin production, and trophic-phase span are controlled by a MYB-related transcription factor in the fungus Ashbya gossypii. Appl. Environ. Microbiol. 2006, 72, 5052–5060. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Liang, Y.; Dai, Y.T.; Yang, C.T.; Duan, M.Z.; Zhang, Z.; Hu, S.N.; Zhang, Z.W.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar] [CrossRef]
- Chen, Y.H.; Yang, X.Y.; He, K.; Liu, M.H.; Li, J.G.; Gao, Z.F.; Lin, Z.Q.; Zhang, Y.F.; Wang, X.X.; Qiu, X.M.; et al. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar] [CrossRef]
- Du, H.; Yang, S.S.; Liang, Z.; Feng, B.R.; Liu, L.; Huang, Y.B.; Tang, Y.X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.; Gong, W.; He, S.; Sun, G.; Sun, J.; Du, X. Genome-wide characterization and expression analysis of MYB transcription factors in Gossypium hirsutum. BMC Genet. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, J.; Hu, R.; Wu, P.; Hou, X.L.; Song, X.M.; Xiong, A.S. Genome-wide analysis of the R2R3-MYB transcription factor genes in Chinese cabbage (Brassica rapa ssp. pekinensis) reveals their stress and hormone responsive patterns. BMC Genom. 2015, 16. [Google Scholar] [CrossRef]
- Ohm, R.A.; de Jong, J.F.; de Bekker, C.; Wösten, H.A.; Lugones, L.G. Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol. Microbiol. 2011, 81, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Sakura, H.; Kanei-Ishii, C.; Nagase, T.; Nakagoshi, H.; Gonda, T.J.; Ishii, S. Delineation of three functional domains of the transcriptional activator encoded by the c-MYB protooncogene. Proc. Natl. Acad. Sci. USA 1989, 86, 5758–5762. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Morikawa, S.; Nakamura, H.; Sekikawa, A.; Inoue, T.; Kanai, H.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structure of a specific DNA complex of the MYB DNA-binding domain with cooperative recognition helices. Cell 1994, 79, 639–648. [Google Scholar] [CrossRef]
- Dias, A.P.; Braun, E.L.; McMullen, M.D.; Grotewold, E. Recently duplicated maize R2R3 MYB genes provide evidence for distinct mechanisms of evolutionary divergence after duplication. Plant Physiol. 2003, 131, 610–620. [Google Scholar] [CrossRef] [PubMed]
- O’Bleness, M.; Searles, V.B.; Dickens, C.M.; Astling, D.; Albracht, D.; Mak, A.C.; Lai, Y.Y.; Lin, C.; Chu, C.; Graves, T.; et al. Finished sequence and assembly of the DUF1220-rich 1q21 region using a haploid human genome. BMC Genom. 2014, 15. [Google Scholar] [CrossRef]
- Luo, M.C.; Deal, K.R.; Murray, A.; Zhu, T.; Hastie, A.R.; Stedman, W.; Sadowski, H.; Saghbini, M. Optical Nano-mapping and analysis of plant genomes. Methods Mol. Biol. 2016, 1429, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Xu, J.; Xiao, S.M.; Liao, B.S.; Gao, Y.; Zhai, C.C.; Qiu, X.H.; Xu, W.; Chen, S.L. Comparative optical genome analysis of two pangolin species: Manis pentadactyla and Manis javanica. Gigascience 2016, 5. [Google Scholar] [CrossRef]
- Xiao, S.; Li, J.; Ma, F.; Fang, L.; Xu, S.; Chen, W.; Wang, Z.Y. Rapid construction of genome map for large yellow croaker (Larimichthys crocea) by the whole-genome mapping in BioNano Genomics Irys system. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Xu, J.; Liu, C.; Zhu, Y.J.; Nelson, D.R.; Zhou, S.G.; Li, C.F.; Wang, L.Z.; Guo, X.; Sun, Y.Z.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Seo, Y.S.; Min, K.; Park, A.R.; Lee, J.; Jin, J.M.; Lin, Y.; Cao, P.; Hong, S.Y.; Kim, E.; et al. A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog. 2011, 7, e1002310. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Gazara, R.K.; Verma, P.K. Transcription factor repertoire of necrotrophic fungal phytopathogen Ascochyta rabiei: Predominance of MYB transcription factors as potential regulators of secretome. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Davidson, C.J.; Tirouvanziam, R.; Herzenberg, L.A.; Lipsick, J.S. Functional evolution of the vertebrate MYB gene family: B-MYB, but neither A-MYB nor c-MYB, complements Drosophila MYB in hemocytes. Genetics 2005, 169, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; He, J.; Dong, J.; Hou, X.; Zhang, X. Genomic survey and expression profiling of the MYB gene family in watermelon. Hortic. Plant J. 2017, 4, 1–15. [Google Scholar] [CrossRef]
- Hou, X.J.; Li, S.B.; Liu, S.R.; Hu, C.G.; Zhang, J.Z. Genome-wide classification and evolutionary and expression analyses of citrus MYB transcription factor families in Sweet Orange. PLoS ONE 2014, 9, e112375. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, X. Genome-wide characterization and expression analysis of MYB transcription factors in Lotus japonicas and Medicago truncatula. Genes Genom. 2017, 39, 831–842. [Google Scholar] [CrossRef]
- Lang, G.; White, J.R.; Argent-Katwala, M.J.; Allinson, C.G.; Weston, K. MYB proteins regulate the expression of diverse target genes. Oncogene 2005, 24, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Law, M.; Holt, C.; Stein, J.C.; Moghe, G.D.; Hufnagel, D.E.; Lei, J.; Achawanantakun, R.; Jiao, D.; Lawrence, C.J. MAKER-P: A tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014, 164, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Chang, H.-Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2014, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S. The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Bio. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | Final Scaffolds | Next-Generation Sequencing Assembly | Bionano Genome Map |
|---|---|---|---|
| Number of scaffolds/maps | 203 | 825 | 246 |
| Total length of scaffolds/maps (Mb) | 34.76 | 36.42 | 33.69 |
| Scaffold/map N50 (kb) | 1109.40 | 432.52 | 138 |
| Gap ratio (%) | 0.91 | 3.37 | - |
| Number of contigs | 570 | 1648 | - |
| Total length of contigs (Mb) | 34.45 | 35.19 | - |
| Contig N50 (kb) | 191.92 | 126.35 | - |
| Repeat (%) | 10.11 | - | - |
| GC content (%) | 50.49 | 50.90 | - |
| Number of protein-coding genes | 10,936 | - | - |
| Average coding gene length (bp) | 1743 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Gao, W.; Wu, X.; Zhao, M.; Qu, J.; Huang, C.; Zhang, J. Genome-Wide Characterization and Expression Analyses of Pleurotus ostreatus MYB Transcription Factors during Developmental Stages and under Heat Stress Based on de novo Sequenced Genome. Int. J. Mol. Sci. 2018, 19, 2052. https://doi.org/10.3390/ijms19072052
Wang L, Gao W, Wu X, Zhao M, Qu J, Huang C, Zhang J. Genome-Wide Characterization and Expression Analyses of Pleurotus ostreatus MYB Transcription Factors during Developmental Stages and under Heat Stress Based on de novo Sequenced Genome. International Journal of Molecular Sciences. 2018; 19(7):2052. https://doi.org/10.3390/ijms19072052
Chicago/Turabian StyleWang, Lining, Wei Gao, Xiangli Wu, Mengran Zhao, Jibin Qu, Chenyang Huang, and Jinxia Zhang. 2018. "Genome-Wide Characterization and Expression Analyses of Pleurotus ostreatus MYB Transcription Factors during Developmental Stages and under Heat Stress Based on de novo Sequenced Genome" International Journal of Molecular Sciences 19, no. 7: 2052. https://doi.org/10.3390/ijms19072052