1. Introduction
The Genus
Impatiens belongs to the family Balsaminaceae, which consists of about 850 species of mostly succulent annual or perennial herbs [
1]. Wild
Impatiens plants are primarily found in the mountainous regions of South-East Asia, south China, India and Africa while some species are also found in Japan, Europe, Russia and North America [
2]. Garden impatiens (
Impatiens walleriana Hook.f.) is one of the most popular flowers in the world, widely grown in garden beds, borders and woodland gardens as bedding plants and in containers, window boxes and hanging baskets as house plants [
3,
4]. Impatiens flowers are available virtually in all colors [
3,
4].
Impatiens downy mildew (IDM), caused by
Plasmopara obducens (J.
Impatiens noli-tangere), is a devastating disease to impatiens and can cause rapid defoliation and plant death. This disease was first reported in Germany in 1877 on
Impatiens noli-tangere, a wild species of
Impatiens native to many temperate countries in the northern hemisphere [
5]. During the 1880s,
P. obducens was identified in North America in native
Impatiens species including
I. capensis (synonym
I. biflora),
I. fulva and
I. pallida [
5]. The first recent occurrence of IDM on
I. walleriana was reported in the UK in 2003 [
6]. Subsequently it spread to other countries in Europe (including Norway (2010), Serbia (2011) and Hungary (2011)) and other continents [
7,
8,
9]. IDM was reported in Taiwan and Japan in 2003 and in Australia in 2008 [
10,
11,
12]. Since then, IDM has been problematic in these countries. In the USA, IDM appeared on
I. walleriana plants in California in 2004 [
13]. In 2011, IDM was reported in eleven states in the USA including Massachusetts, New York and Minnesota [
14]. By 2012, this disease spread to 33 states [
13]. In 2013, IDM was reported in Hawaii [
15]. Outbreak of this disease in the USA in this short period of time has caused the loss of hundreds of millions of dollars, reducing the national annual wholesale value of impatiens from approximately
$150 million in 2005 to
$65 million in 2015 [
16]. While
I. walleriana is highly susceptible to IDM, New Guinea Impatiens (NGI) (
Impatiens hawkeri) and its interspecific hybrids have shown resistance to IDM [
17]. NGI has been grown as a substitute of garden impatiens currently and occupies a
$55-million wholesale market [
16]. Although the market of NGI is increasing, it ranked third among the alternatives of garden impatiens [
18].
There has been a strong interest in introducing IDM resistance into
I.
walleriana cultivars due to continued strong consumer demand.
Impatiens walleriana cultivars are available in a wide array of vibrant colors, possess excellent shade tolerance and are well adapted to a wide range of growing conditions in containers and garden beds [
3]. Moreover, the ease of culturing, wide and greater availability of the seeds and cutting materials has increased the demand of garden impatiens [
3]. Use of traditional breeding approaches to transferring the resistance from NGI to garden impatiens has been impeded by the differences between NGI and garden impatiens in chromosome number (2
n = 2
× = 16 in
I.
walleriana and 2
n = 2
× = 32 in NGI), size and morphology [
19]. Hence, it is necessary to identify and isolate the gene(s) conferring IDM resistance in NGI and transfer the identified gene(s) into garden impatiens.
Disease resistance (
R) genes are the most important component of the plant defense mechanism to confer resistance to pathogens carrying matching avirulence genes [
20]. A great majority of isolated plant
R genes encode nucleotide-binding site leucine-rich repeats (NB-LRR) proteins [
21]. Based on structure, NB-LRR proteins are separated into Toll/interleukin-1 receptor (TIR)-domain-containing (TNL) and coiled coil (CC)-domain-containing (CNL) subfamilies. Plant NB-LRR proteins function in a network through signaling pathways and induce a series of defense responses such as initiation of oxidative burst, calcium and ion fluxes, mitogen-associated protein kinase cascade, induction of pathogenesis-related genes and hypersensitive responses [
22,
23,
24,
25]. Recent studies have shown that the TNL subfamily R proteins transduce signals through the enhanced disease susceptibility 1 (EDS1) protein and the CNL subfamily R proteins do so through non-race specific disease resistance 1 (NDR1) protein with some interchanges occurring in different plants [
26]. There also exists a separate independent signal transduction pathway activated by Arabidopsis CNL proteins RPP8 and RPP13.
Few genomic resources are available in impatiens. Genomic and transcriptomic data are needed in impatiens to facilitate the identification of gene(s) conferring IDM resistance in NGI and the development of simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers for impatiens breeding. With the rapid development of next generation sequencing and analytical tools, whole-genome and transcriptome sequencing has become possible in non-model organisms. Transcriptome sequencing and characterization can provide a descriptive and biological insight of the functionality of an organism. Recently, transcriptome sequencing has been used commonly to identify candidate
R genes and to develop molecular markers for disease resistance breeding [
27,
28]. There has been very few or no study on genes involved in disease resistance using transcriptome sequencing in impatiens. Here, we report the sequencing, assembly, annotation and characterization of the leaf transcriptomes of two impatiens cultivars (Super Elfin
® XP Pink (SEP) and SunPatiens
® Compact Royal Magenta (SPR)) that differ in IDM resistance and the use of the transcriptome data to identify differentially expressed transcripts and candidate
R genes and SSR and SNP sites for development of molecular markers. Results of this study, including the assembled transcriptomes and the identified candidate genes and polymorphic sites, will greatly facilitate the development of molecular markers for dissecting the resistance mechanism for downy mildew resistance in NGI, tagging the responsible gene loci and expediting the improvement of downy mildew resistance in impatiens.
3. Discussion
RNA-Seq has become a very powerful technique for analysis of gene expression, identification of candidate genes involved in the expression of important traits and rapid discovery of large numbers of SSRs and SNPs in non-model plant species [
29,
30,
31]. In this study, the leaf transcriptomes of impatiens were sequenced, assembled, annotated and compared to identify candidate genes potentially involved in resistance against IDM. We found fifteen unigenes that were more than 2000 nucleotides long and had at least 2-fold more transcripts in the IDM-resistant cultivar SPR than in the IDM-susceptible cultivar SEP. Most of the identified candidate genes showed high levels of similarity to the NB-LRR or LRR gene families. Members of the NB-LRR gene family are known to confer plants resistance to diseases caused by bacteria, fungi and viruses and are localized in the cytoplasm [
32,
33,
34,
35]. LRR genes have been shown to be involved in fungal resistance. The LRR domain in the NB-LRR and LRR
R-proteins plays a critical role in defense response by perceiving signals from effectors released by pathogens or pathogens themselves and transducing the signals to initiate the defense responses [
36,
37,
38,
39]. The NB-LRR proteins are divided into two groups based on the presence of Toll/interleukin-1 receptor (TIR) or coiled-coil (CC) domain in the amino terminal end. Despite the common function of pathogen recognition, the sequence and signaling mechanism of TNL and CNL proteins are different [
40,
41,
42]. In this study, there were two genes identified as TNLs or CNL encoding proteins.
RPP13 is a part of a NB-ARC domain known for the presence in APAF-1 (apoptopic protease-activating factor-1), R proteins and CED-4 (
Caenorhabditis elegans death-4 protein) and belongs to the CC-NBS-LRR family that encodes NBS-LRR type R protein with a putative amino-terminal leucine zipper [
43].
RPP13 has been reported to confer resistance to five isolates of
Peronospora parasitica that caused downy mildew in
A. thaliana [
44]. More than 20 loci for
RPP genes has been already identified in
A. thaliana conferring recognition of
P. parasitica [
45]; genes from three loci,
RPP1,
RPP5 and
RPP8 have been cloned [
46,
47,
48].
RPP13 has been placed in the subclass of NB-LRR type
R proteins that have putative leucine zipper (LZ) domain located near the N-terminus [
49]. Before it was known that these loci confer resistance to downy mildew in
A. thaliana, RPP13 was only known to possess resistant activity to
Pseudomonas syringae pathovars [
36,
37,
38,
39]. After the understanding of TIR type NB-LRR
R genes found at the complex
RPP1 and
RPP5 loci [
47,
48,
49]. In
Arabidopsis, RPP13 has been known to be reminiscent of
R genes
RPM1 [
42] and
RPS2 [
36,
38]. A recent study shows functionally diverged alleles as a distinction in
RPP13 at a simple
R gene locus [
44]. In this study, there were two putative disease resistance RPP13-proteins identified in the IDM-resistant cultivar with higher amount than in the susceptible cultivar. Presence of genes similar to
RPP13 indicate the possibility of
RPP-like genes conferring the resistance to IDM. Resistance to
P. parasitica by
RPP13 in
A. thaliana Niederzenz accession is known to be independent to either Salicylic acid,
NRD1 or
EDS1 [
50] triggered by the avirulence gene
ATR13 [
51].
RPP13 encodes protein that is predicted to be located in cytoplasm [
43]. In addition to
RPP-like genes, increased expression of other genes similar to LETM1-like protein, Cation efflux family protein, Receptor like protein 35 and DNA-binding storekeeper protein-related transcriptional regulator indicates the complexity of defense mechanism in the resistant line to IDM. Further research on the mode of resistance in New Guinea Impatiens for IDM could reveal the involvement of these proteins in conjunction with RPP13 to confer resistance or exhibit different mode of action.
Functional annotation of these genes also grouped them into different categories on the basis of similarity. The common method of recognition and signaling mechanisms for resistance exhibited by different classes of gene families differently in unrelated group of pathogens indicates the possibility of functional diversity of R genes to defend the host from different kinds of pathogens in different species of host. It has been previously reported that R genes exhibit polymorphism regarding the role in defense mechanism. There were other unigenes that were identified to be functionally similar to RPS2-like proteins family in the transcriptome but not all of them were significantly upregulated in the resistant cultivar in this experiment.
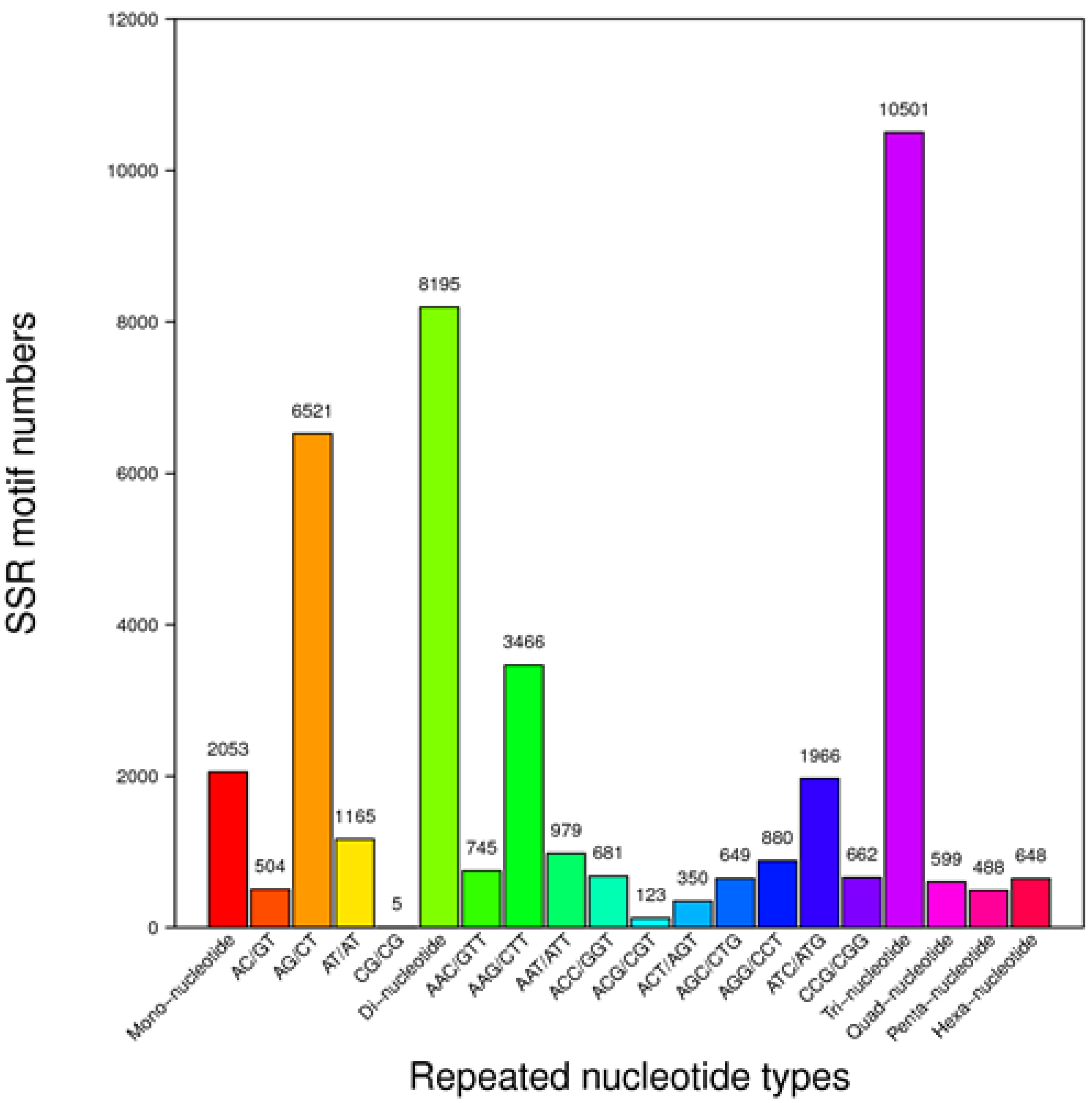
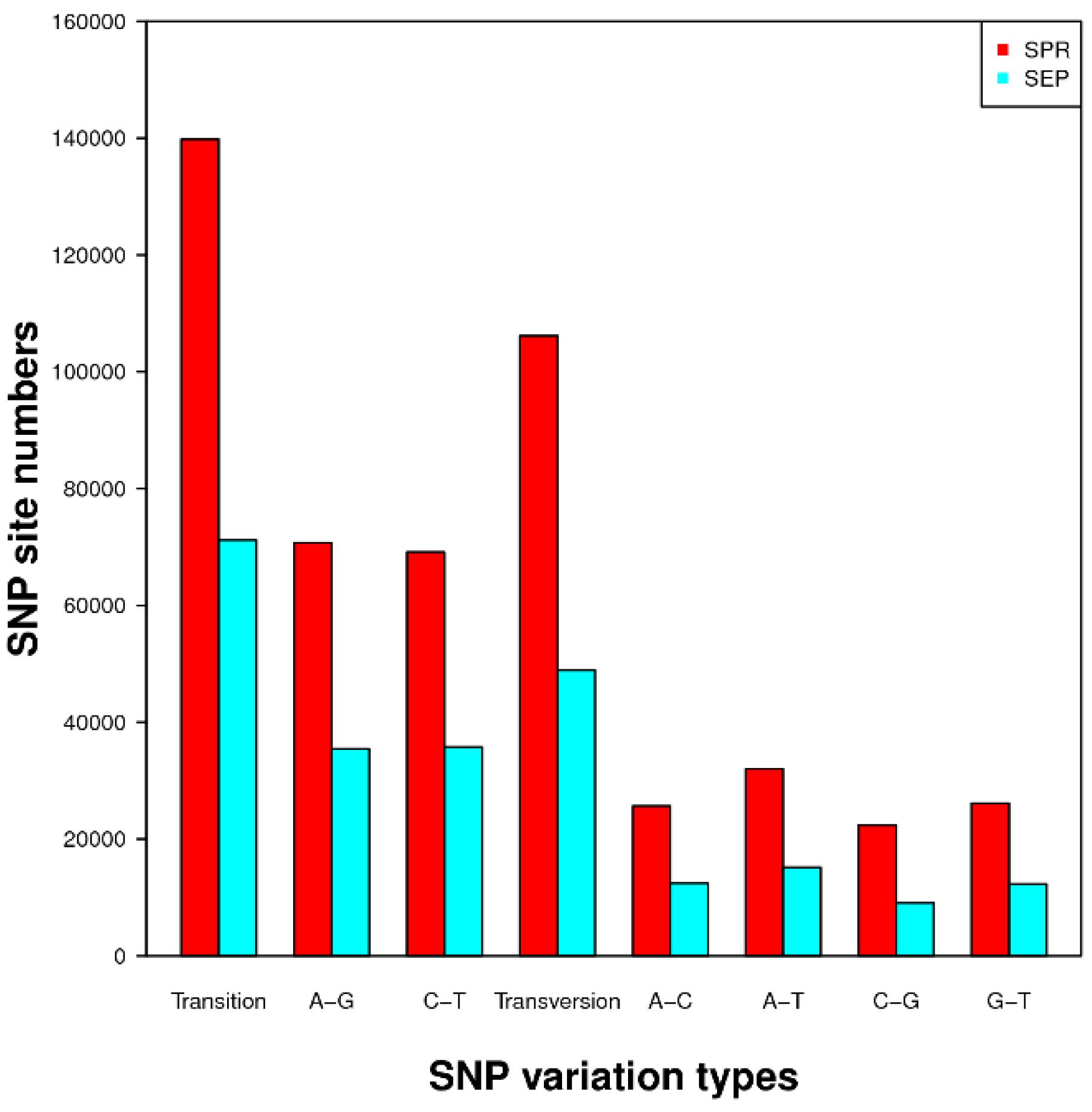
Being abundant and randomly distributed in plant genomes, SNPs and SSRs are very useful loci for development of molecular markers for high-density, high-resolution gene or genome mapping and marker-assisted breeding. RNA-Seq and bioinformatics analytical tools have made the discovery of SSR and SNP sites easy and fast. So far, molecular markers have not been reported for downy mildew resistance in impatiens. This study resulted in the discovery of 22,484 SSRs and 245,936 and 120,073 SNPs in SPR and SEP, respectively. These SSR and SNP sites offer a very useful resource for developing molecular markers that can be used by geneticists and breeders for genetic studies and improvement of impatiens.
4. Materials and Methods
4.1. Plant Materials
IDM-susceptible cultivar Super Elfin® XP Pink (SEP, I. walleriana) and IDM-resistant cultivar SunPatiens® (SPR, I. hawkeri) were used in this study. Three plants of each cultivar were grown individually in 15-cm plastic containers filled with the commercial soilless potting mixture Faffard® 3B (50% Canadian peat and 50% of the mixture of vermiculite, pine bark and perlite) (Agawam, MA, USA) in a greenhouse facility at the University of Florida’s Gulf Coast Research and Education Center (UF/GCREC), Wimauma, FL, USA. Plants were watered as needed and no fungal pesticides were applied. Mature leaves from both resistant and susceptible plants were sampled and they were instantly frozen in liquid nitrogen and stored in a deep freezer at −80 °C until use.
4.2. RNA Isolation, cDNA Synthesis and Sequencing
Frozen leaf tissue samples of SEP and SPR were shipped on dry ice to Beijing Genomics Institute (BGI) in Shenzhen, China. Total RNA was extracted from the impatiens leaf tissues using pBiozol Total RNA Extraction Reagent (BioFlux, Hangzhou, China) following manufacturer’s instructions. The RNA concentration, RNA integrity number (RIN) and the 28S/18S ratio were determined on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). An equal amount of RNA from each of the three biological replicates was combined to form a pool of resistant and susceptible RNA samples for mRNA isolation and cDNA synthesis. The combined RNA samples were treated with DNase I to remove residual genomic DNA and mRNA was isolated using magnetic beads with Oligo (dT). The isolated mRNA molecules were fragmented using the Elute Prime Fragment Mix from Illumina TruSeqTM RNA sample prep kit v2 (Illumina, San Diego, CA, USA) at 94 °C for 8 min. The fragmented mRNAs were used as templates for cDNA synthesis. First strand cDNA was synthesized by using the First Strand Master Mix and SuperScript II from Invitrogen (Carlsbad, CA, USA) and amplified at 25 °C for 10 min, 42 °C for 50 min and 70 °C for 15 min. Second cDNA strand was synthesized using the Second Strand Master Mix from Invitrogen at 16 °C for 1 h. The resulted cDNA fragments were purified, their ends were repaired using the End Repair Master Mix from Invitrogen at 30 °C for 30 min and the Ampure XP beads (Beckman Coulter, Brea, CA, USA) and a single adenine (A) base were added to each end of the cDNA fragments. End-repaired and A-added short fragments were joined with adapters. The adapter-ligated cDNA was PCR-amplified to generate sequencing libraries. The libraries were analyzed with an Agilent 2100 Bioanalyzer and the ABI StepOnePlus Real-Time PCR System for quantifying and qualifying the sample library. Sequencing of cDNA was performed on an Illumina HiSeq™ 2000, generating 100 bp paired-end reads.
4.3. Sequence Filtering, De Novo Assembly and Transcriptome Analysis
Raw sequence reads were cleaned by removing the adapters attached at the ends and reads with unknown nucleotides more than 5% were removed using BGI’s internal software (filter_fq). Low quality reads with more than 20% of quality value < 10 were removed, Q20 percentage, proportion of unknown nucleotides in clean reads (N) percentage and GC percentage among total nucleotides were calculated and the remaining clean reads were used in further analysis. De novo transcriptome assembly was done using the Trinity software (
http://trinityrnaseq.sourceforge.net/) [
52] with the following parameters: minimum contig length of 100, minimum glue 3, group pair distance 250, path reinforcement distance 85 and minimum kmer coverage 3 using the cleaned short reads. The longest, non-redundant, unique transcripts were defined as unigenes. Further processing of the sequences for redundancy removal and splicing was done using the sequence clustering software, TIGR Gene Indices Clustering Tools (TGICL) v2.1 (
http://sourceforge.net/projects/tgicl/files/tgicl%20v2.1/) with minimum overlap length 40, base quality cutoff of clipping 10 and maximum length of unmatched overhangs 20. Homologous transcript clustering was done using Phrap release 23.0 (
http://www.phrap.org/) with repeat stringency 0.95, minimum match 35 and minimum score 35.
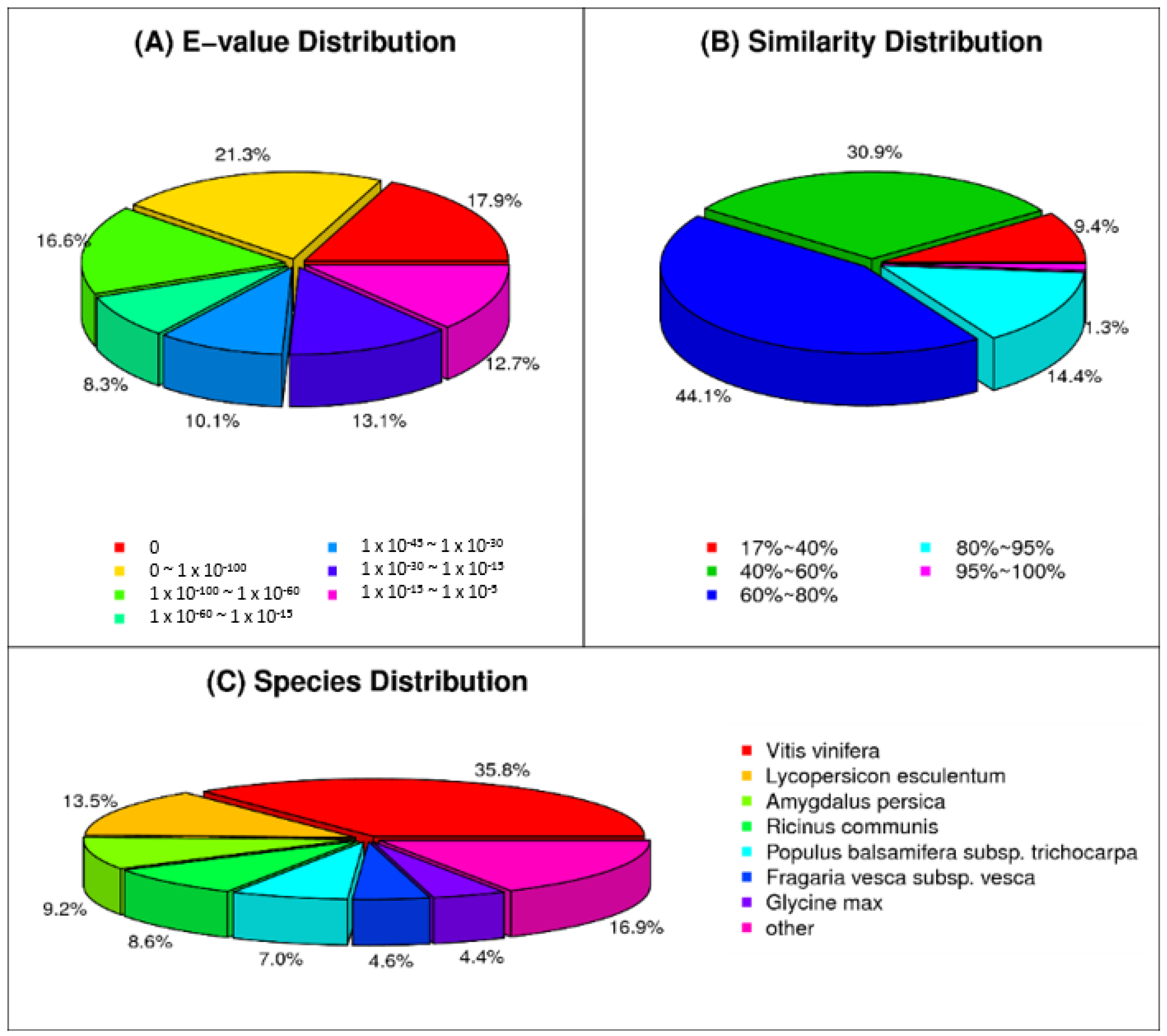
Functional annotation of the unigenes was done by comparing them using the NR (non-redundant protein) and NT (non-redundant nucleotide) databases of National Center for Biotechnology Information (
http://blast.ncbi.nlm.nih.gov/Blast.cgi), SWISS-PROT database (European Bioinformatics Institute,
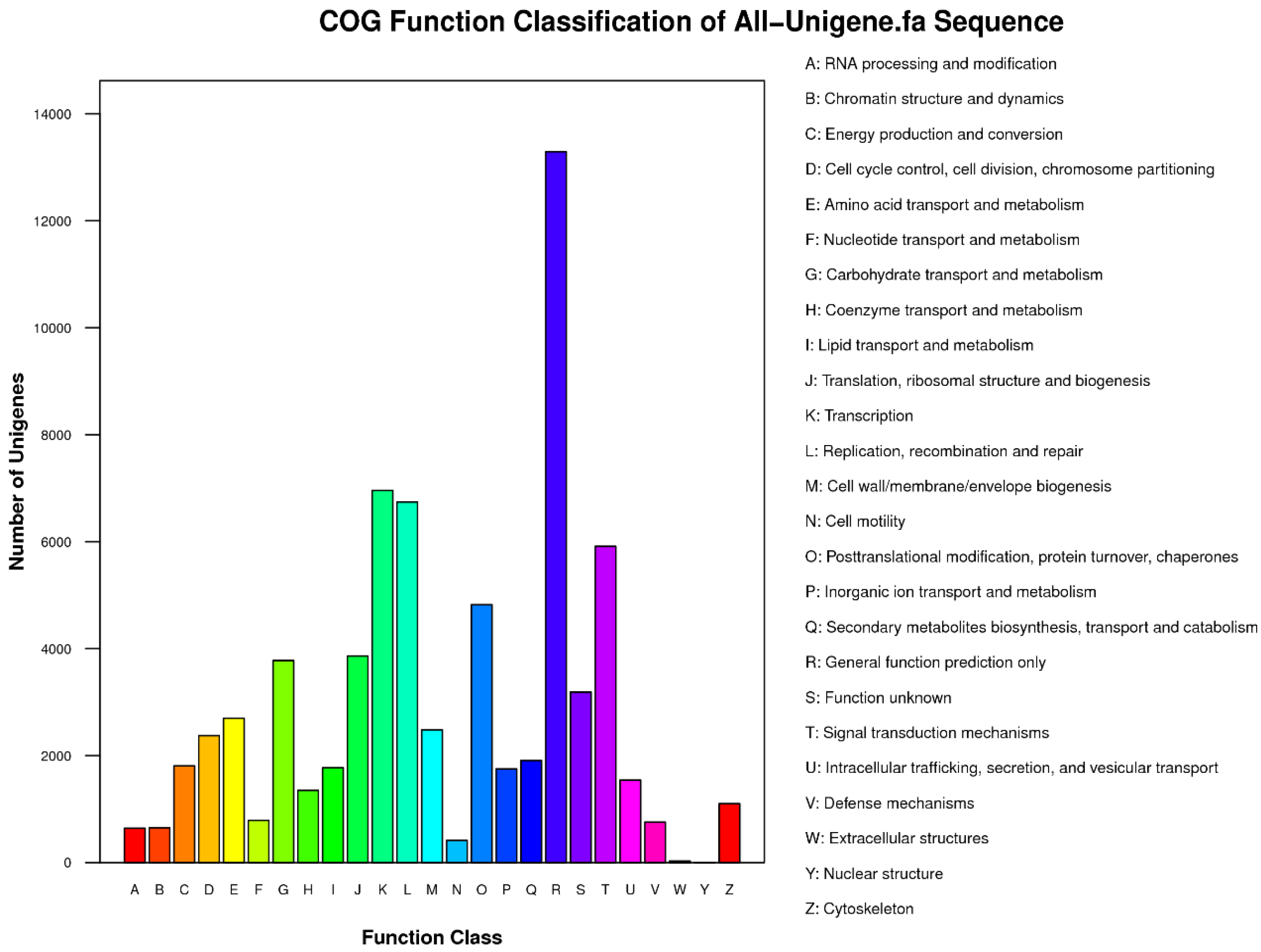
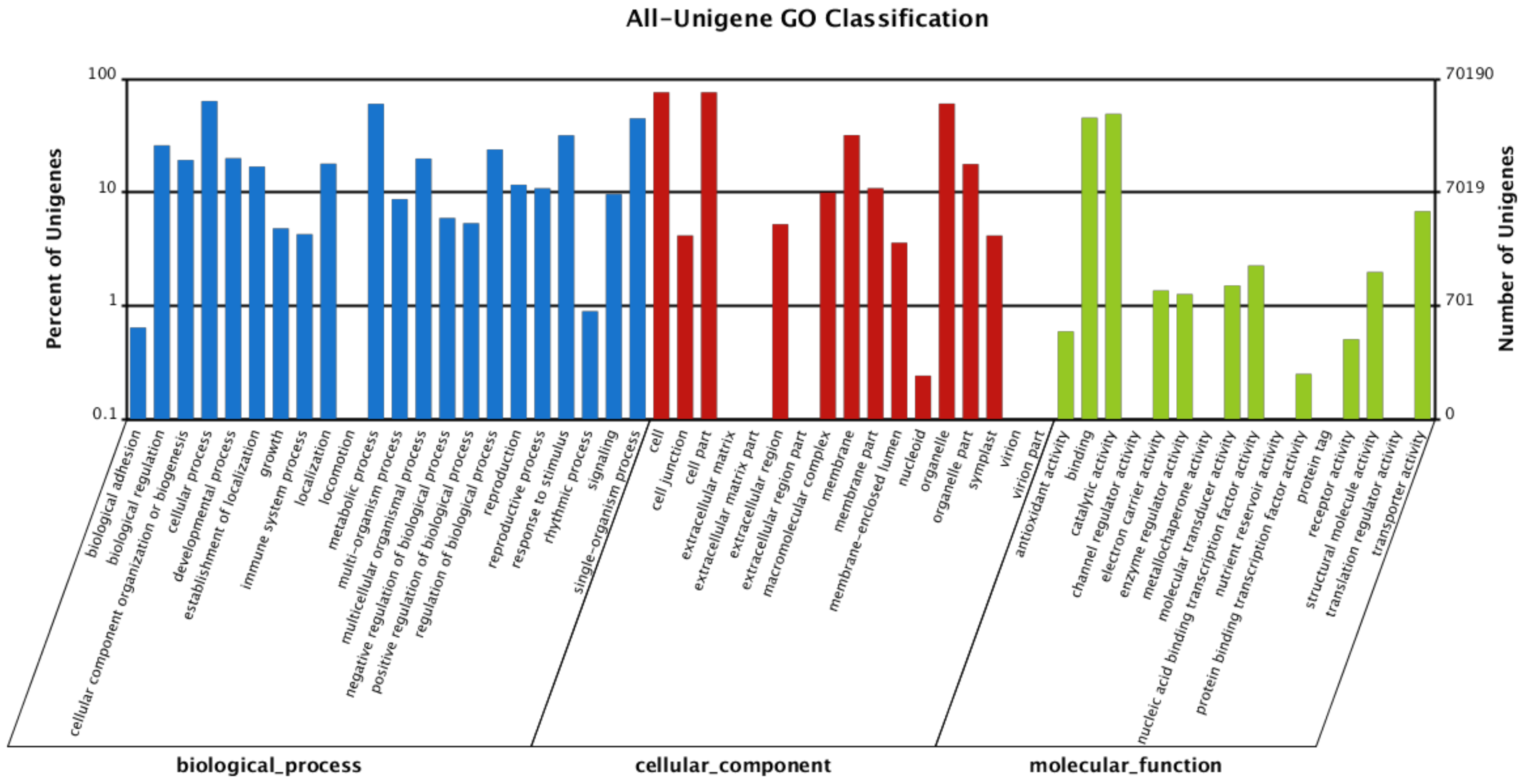
ftp://ftp.ebi.ac.uk/pub/databases/swissprot/), KEGG (Kyoto Encyclopedia of Genes and Genomes database), COG (Clusters of Orthologous Groups of proteins database) and GO (Gene Ontology) with BLASTx using
E-value cutoff of 1 × 10
−5. The best aligning results were used to determine the sequence direction of unigenes. When the results of databases conflicted each other, a priority order of NR, SWISS-PROT, KEGG and COG was followed to determine the sequence direction. When unigenes were found to be unaligned with any of the databases, ESTScan was used to determine the sequence direction [
53]. Blast2Go (
http://www.blast2go.com/b2ghome) was used to annotate unigenes generated by NR annotation to get GO annotation [
54]. After GO annotation, WEGO software was used for functional classification of the unigenes and to understand the distribution of gene functions from the macro level [
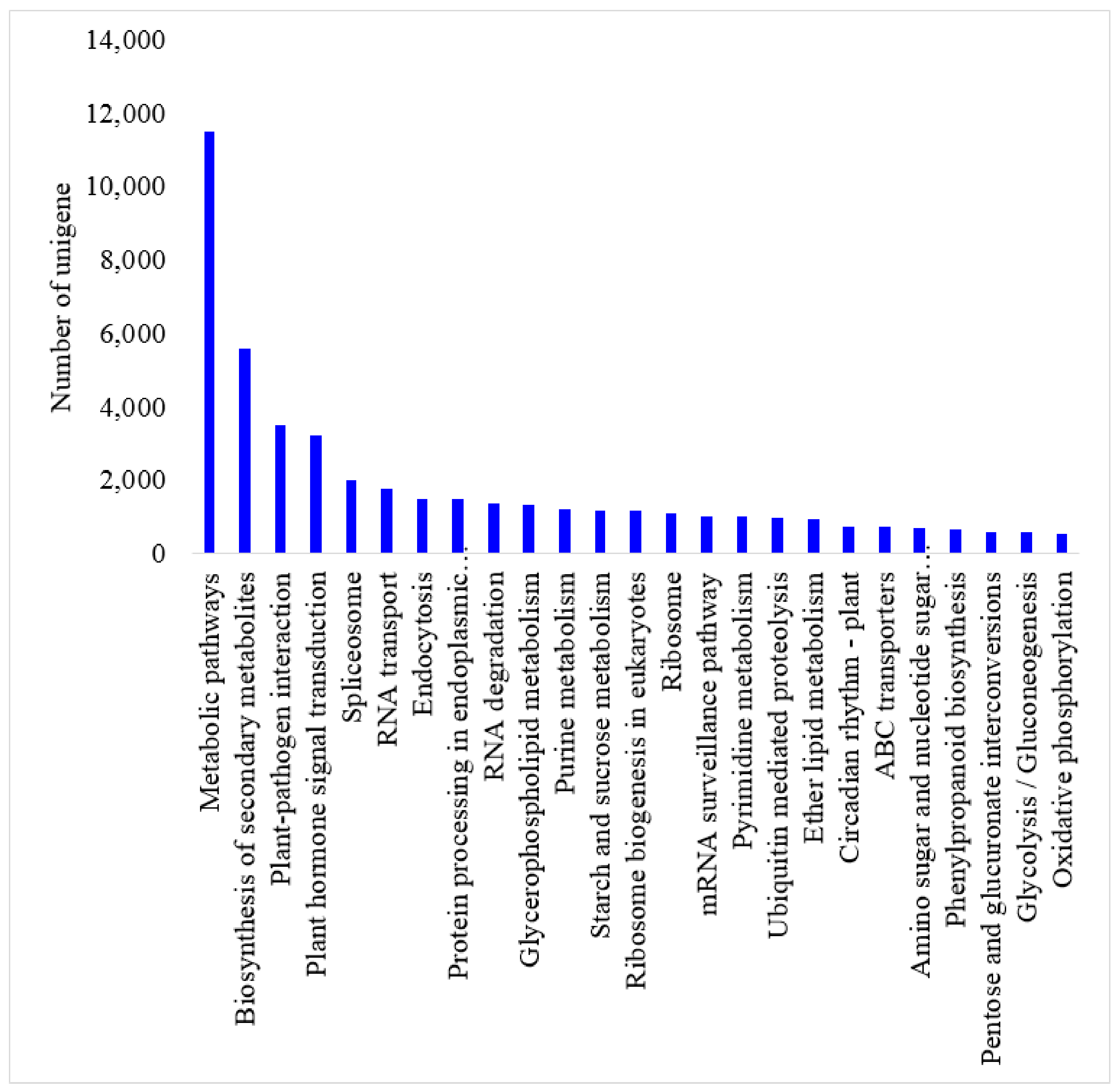
55]. KEGG database was used for metabolic pathway analysis of gene products in cell and function of gene products. Further study of unigene pathways was done using Path_finder (
http://www.genome.jp/) with default parameters [
56].
4.4. Unigenes Sequence Comparison and Phylogenetic Analysis
The sequences of selected unigenes from impatiens were compared to the Arabidopsis Information Resource (TAIR) Database (
https://www.arabidopsis.org/) [
57] to show the functional similarity and the best aligning results were used. BLAST was done using CDS sequences of fifteen unigenes (
Table 6) involved in disease resistance to the nr/nt database for sequence similarity.
4.5. Identification of SSRs and SNPs
SSRs in the unigene sequences were detected using Microsatellite (MISA) (
http://pgrc.ipk-gatersleben.de/misa/misa.html). The minimum cut-off values for the identification of mono-, di-, tri, tetra-, penta- and hexa-nucleotide SSRs were 12, 6, 5, 5, 4 and 4, respectively. Only SSRs with 150-bp flanking sequences on both ends in the unigenes were retained. SSR primers were designed using the Primer3 software (Release 2.3.4,
http://www.onlinedown.net/soft/51549.htm) and the default parameters [
58]. The SOAPsnp software Release 1.03 (Short Oligonucleotide Analysis Package single nucleotide polymorphism) (
http://soap.genomics.org.cn/soapsnp.html) was used to identify SNPs by aligning consensus sequences from each cultivar to the transcriptome assembly [
59].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}