The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers

 and
and
Abstract
:
1. Introduction
2. Results
2.1. Clinical Phenotypes and “Biochemical” Phenotypes of Phosphomannomutase 2 (PMM2) Mutants Do Not Correlate
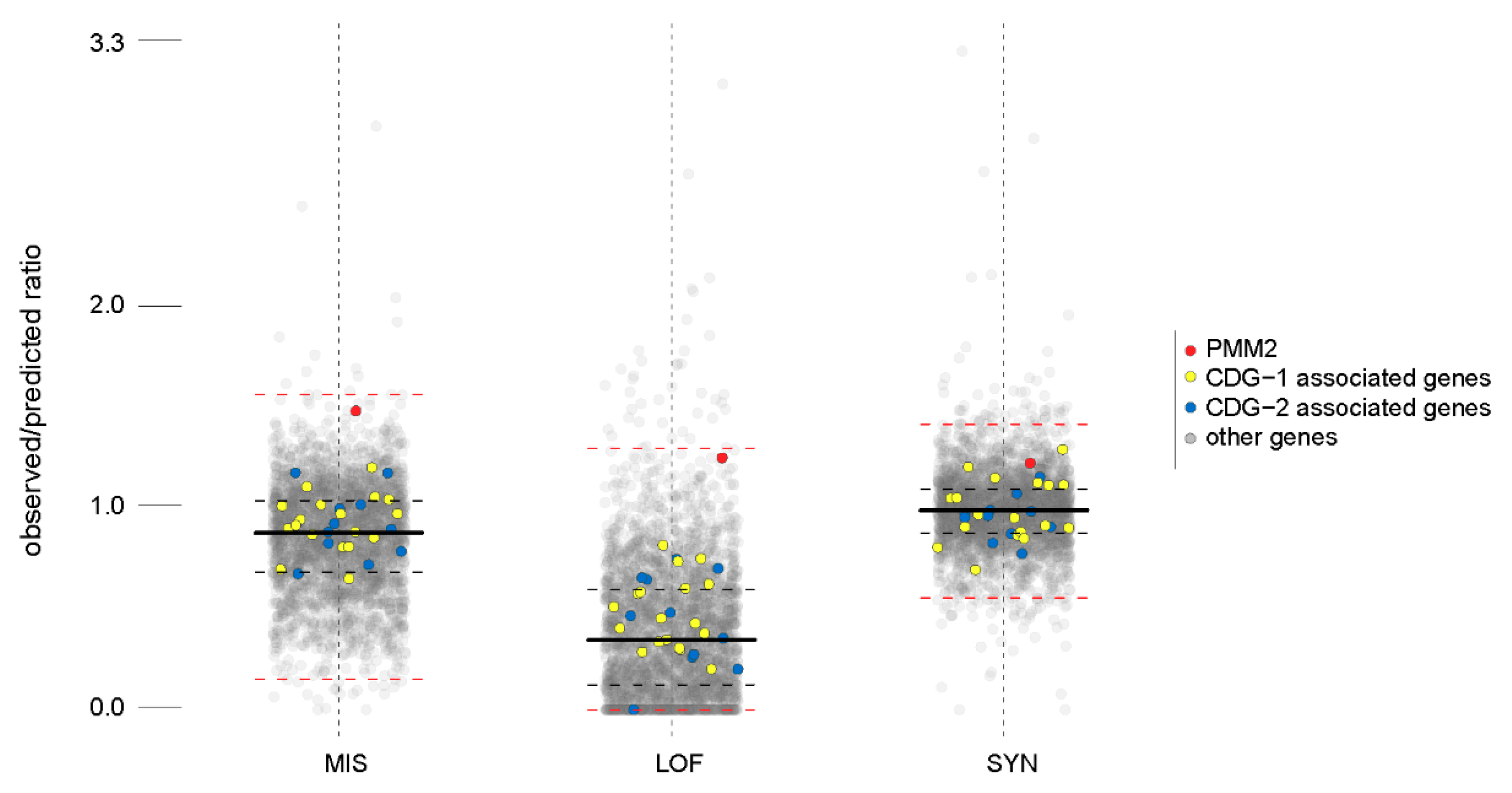
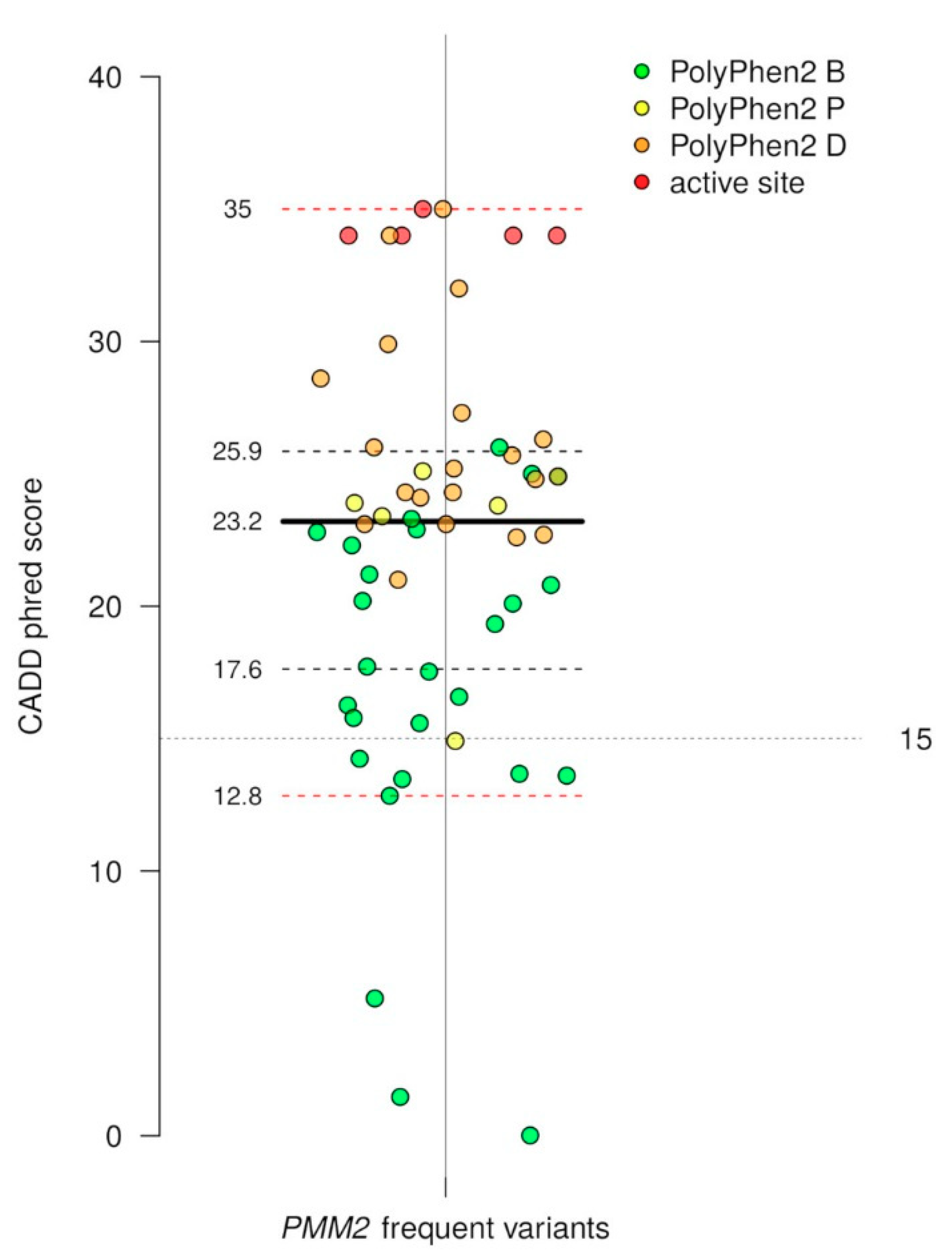
2.2. Missense and Loss of Function Mutations in PMM2 Are Unexpectedly Frequent in the General Population
2.3. Genes Involved in Congenital Disorders of Glycosylation I (CDG-I) Pathologies Have Frequent Variants in the General Population
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Enzymatic Assays
4.3. Thermal Stability
4.4. Miscellaneous
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| M1P | mannose-1phosphate |
| M16 | mannose-1,6-bisphosphate |
| G1P | glucose-1phosphate |
| G16 | glucose-1,6-bisphosphate |
References
- Pirard, M.; Collet, J.F.; Matthijs, G.; Van Schaftingen, E. Comparison of PMM1 with the phosphomannomutases expressed in rat liver and in human cells. FEBS Lett. 1997, 411, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Veiga-da-Cunha, M.; Vleugels, W.; Maliekal, P.; Matthijs, G.; van Schaftingen, E. Mammalian phosphomannomutase PMM1 is the brain IMP-sensitive glucose-1,6-bisphosphatase. J. Biol. Chem. 2008, 283, 33988–33993. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Liguori, L.; Viscido, G.; Cubellis, M.V.; Andreotti, G. A mutant of phosphomannomutase1 retains full enzymatic activity, but is not activated by IMP: Possible implications for the disease PMM2-CDG. PLoS ONE 2017, 12, e0189629. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim. Biophys. Acta 2009, 1792, 827–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, H.H.; Miller, K.L.; Harbison, M.D.; Slonim, A.E. Multiple serum protein abnormalities in carbohydrate-deficient glycoprotein syndrome: Pathognomonic finding of two-dimensional electrophoresis? Clin. Chem. 1992, 38, 1390–1392. [Google Scholar] [PubMed]
- Henry, H.; Tissot, J.D.; Messerli, B.; Markert, M.; Muntau, A.; Skladal, D.; Sperl, W.; Jaeken, J.; Weidinger, S.; Heyne, K.; et al. Microheterogeneity of serum glycoproteins and their liver precursors in patients with carbohydrate-deficient glycoprotein syndrome type I: Apparent deficiencies in clusterin and serum amyloid p. J. Lab. Clin. Med. 1997, 129, 412–421. [Google Scholar] [CrossRef]
- Kjaergaard, S.; Skovby, F.; Schwartz, M. Carbohydrate-deficient glycoprotein syndrome type 1a: Expression and characterisation of wild type and mutant PMM2 in E. coli. Eur. J. Hum. Genet. 1999, 7, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G. Congenital disorders of glycosylation. Trends Biochem. Sci. 2000, 25, 428. [Google Scholar] [CrossRef]
- Matthijs, G.; Schollen, E.; Heykants, L.; Grunewald, S. Phosphomannomutase deficiency: The molecular basis of the classical jaeken syndrome (CDGS type IA). Mol. Genet. Metab. 1999, 68, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Pirard, M.; Matthijs, G.; Heykants, L.; Schollen, E.; Grunewald, S.; Jaeken, J.; van Schaftingen, E. Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Lett. 1999, 452, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Le Bizec, C.; Vuillaumier-Barrot, S.; Barnier, A.; Dupre, T.; Durand, G.; Seta, N. A new insight into PMM2 mutations in the french population. Hum. Mutat. 2005, 25, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.I.; Perez-Cerda, C.; Abia, D.; Gamez, A.; Briones, P.; Artuch, R.; Desviat, L.R.; Ugarte, M.; Perez, B. Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): Expression analysis of PMM2-CDG mutations. J. Inherit. Metab. Dis. 2011, 34, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Pedone, E.; Giordano, A.; Cubellis, M.V. Biochemical phenotype of a common disease-causing mutation and a possible therapeutic approach for the phosphomannomutase 2-associated disorder of glycosylation. Mol. Genet. Genom. Med. 2013, 1, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreotti, G.; Monti, M.C.; Citro, V.; Cubellis, M.V. Heterodimerization of two pathological mutants enhances the activity of human phosphomannomutase2. PLoS ONE 2015, 10, e0139882. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Carrozzi, M.; Parini, R.; Battini, R.; Martinelli, D.; Elia, M.; Spada, M.; Lilliu, F.; Ciana, G.; Burlina, A.; et al. A nationwide survey of PMM2-CDG in italy: High frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2015, 262, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Monin, M.L.; Mignot, C.; de Lonlay, P.; Heron, B.; Masurel, A.; Mathieu-Dramard, M.; Lenaerts, C.; Thauvin, C.; Gerard, M.; Roze, E.; et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: Outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J. Rare Dis. 2014, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, S.; Schollen, E.; van Schaftingen, E.; Jaeken, J.; Matthijs, G. High residual activity of PMM2 in patients’ fibroblasts: Possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am. J. Hum. Genet. 2001, 68, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Vuillaumier-Barrot, S.; Hetet, G.; Barnier, A.; Dupre, T.; Cuer, M.; de Lonlay, P.; Cormier-Daire, V.; Durand, G.; Grandchamp, B.; Seta, N. Identification of four novel PMM2 mutations in congenital disorders of glycosylation (CDG) ia french patients. J. Med. Genet. 2000, 37, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Schollen, E.; van Schaftingen, E.; Cassiman, J.J.; Jaeken, J. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1a. Am. J. Hum. Genet. 1998, 62, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Robertson, A.D. Some thermodynamic implications for the thermostability of proteins. Protein Sci. 2001, 10, 1187–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, S.; Schwartz, M.; Skovby, F. Congenital disorder of glycosylation type Ia (CDG-Ia): Phenotypic spectrum of the R141H/F119l genotype. Arch. Dis. Child. 2001, 85, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Quental, R.; Moleirinho, A.; Azevedo, L.; Amorim, A. Evolutionary history and functional diversification of phosphomannomutase genes. J. Mol. Evol. 2010, 71, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blekhman, R.; Man, O.; Herrmann, L.; Boyko, A.R.; Indap, A.; Kosiol, C.; Bustamante, C.D.; Teshima, K.M.; Przeworski, M. Natural selection on genes that underlie human disease susceptibility. Curr. Biol. 2008, 18, 883–889. [Google Scholar] [CrossRef] [PubMed]
- ExAC. Available online: http://exac.broadinstitute.org/ (accessed on 15 March 2018).
- HGMD. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 15 March 2018).
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Sift: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous snvs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Briones, P.; Vilaseca, M.A.; Schollen, E.; Ferrer, I.; Maties, M.; Busquets, C.; Artuch, R.; Gort, L.; Marco, M.; van Schaftingen, E.; et al. Biochemical and molecular studies in 26 spanish patients with congenital disorder of glycosylation type Ia. J. Inherit. Metab. Dis. 2002, 25, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.I.; Perez-Cerda, C.; Desviat, L.R.; Matthijs, G.; Ugarte, M.; Perez, B. Functional analysis of three splicing mutations identified in the PMM2 gene: Toward a new therapy for congenital disorder of glycosylation type Ia. Hum. Mutat. 2009, 30, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, F.; Worthington, V.; Champion, M.; Beesley, C.; Charlwood, J.; Clayton, P.; Keir, G.; Mian, N.; Winchester, B. Genotypes and phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. J. Inherit. Metab. Dis. 2000, 23, 162–174. [Google Scholar] [CrossRef] [PubMed]
- de Lonlay, P.; Seta, N.; Barrot, S.; Chabrol, B.; Drouin, V.; Gabriel, B.M.; Journel, H.; Kretz, M.; Laurent, J.; Le Merrer, M.; et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: A series of 26 cases. J. Med. Genet. 2001, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Freeze, H.H.; Westphal, V. Balancing N-linked glycosylation to avoid disease. Biochimie 2001, 83, 791–799. [Google Scholar] [CrossRef]
- Febbraio, M.; Guy, E.; Silverstein, R.L. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dai, X.; Liu, Y.; Li, J.; Liu, Z.; Yin, P.; Chen, J.; Wang, N.; Zhang, P. Mtus1 silencing promotes e-selectin production through p38 MAPK-dependent CREB ubiquitination in endothelial cells. J. Mol. Cell. Cardiol. 2016, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carrera, N.; Arrojo, M.; Paz, E.; Ramos-Rios, R.; Agra, S.; Paramo, M.; Brenlla, J.; Costas, J. Testing the antagonistic pleiotropy model of schizophrenia susceptibility by analysis of DAOA, PPP1R1B, and APOL1 genes. Psychiatry Res. 2010, 179, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Giurgea, I.; Michel, A.; Le Merrer, M.; Seta, N.; de Lonlay, P. Underdiagnosis of mild congenital disorders of glycosylation type Ia. Pediatr. Neurol. 2005, 32, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Kjaergaard, S.; Schollen, E.; Martens, K.; Grunewald, S.; Schwartz, M.; Matthijs, G.; Freeze, H.H. A frequent mild mutation in ALG6 may exacerbate the clinical severity of patients with congenital disorder of glycosylation ia (CDG-Ia) caused by phosphomannomutase deficiency. Hum. Mol. Genet. 2002, 11, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Bortot, B.; Cosentini, D.; Faletra, F.; Biffi, S.; De Martino, E.; Carrozzi, M.; Severini, G.M. PMM2-CDG: Phenotype and genotype in four affected family members. Gene 2013, 531, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Sabry, S.; Vuillaumier-Barrot, S.; Mintet, E.; Fasseu, M.; Valayannopoulos, V.; Heron, D.; Dorison, N.; Mignot, C.; Seta, N.; Chantret, I.; et al. A case of fatal type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Vuillaumier-Barrot, S.; Le Bizec, C.; Durand, G.; Seta, N. The T911C (F304S) substitution in the human alg6 gene is a common polymorphism and not a causal mutation of CDG-Ic. J. Hum. Genet. 2001, 46, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Pirard, M.; Achouri, Y.; Collet, J.F.; Schollen, E.; Matthijs, G.; Van Schaftingen, E. Kinetic properties and tissular distribution of mammalian phosphomannomutase isozymes. Biochem. J. 1999, 339, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2013. [Google Scholar]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Identification and analysis of conserved pockets on protein surfaces. BMC Bioinform. 2013, 14 (Suppl. 7), S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wang, K. Wannovar: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [PubMed]
- ANNOVAR. Annovar Users’ Guide. Available online: http://annovar.openbioinformatics.org/en/latest/user-guide/filter/ (accessed on 15 March 2018).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PMM2 | Relative Activity | Km M1P (µM) | Experimental Conditions for Km M1P | Km M16 (µM) | Experimental Conditions for Km M16 | Long-Term Stability Residual Activity after 10 min @ 40 °C | Melting Temperature (°C) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Wild-type | 100 | 18 | 10 µM M16 | 1.1 | 200 µM M1P | 100 | - | [10] |
| 100 | 16.0 ± 1.5 | 3 µM M16 | 5.4 ± 0.6 * | * 200 µM M1P | 100 ** 100 (in the presence of 200 µM G16) ** | 53.5 ± 0.9 *** | [13] * [3] ** this paper *** [21] | |
| Phe119Leu | 24.5 | 45 | 50 µM M16 | 4.9 | 200 µM M1P | ~60 | - | [10] |
| 29 | 11.8 ± 1.0 | 3 µM M16 | nd | nd | ~95 (37 °C) | 45.0 ± 0.8 *** | [13] *** [21] | |
| Val29Met | 50 | 21 | 10 µM M16 | 1.1 | 200 µM M1P | ~55 | - | [10] |
| 89 | 33.0 ± 3.9 | 3 µM M16 | 3.7 ± 0.5 | 200 µM M1P | 83 100 (in the presence of 200 µM G16) | 48.1 ± 1.0 | this paper | |
| Val231Met | 38.5 | 18 | 10 µM M16 | 1.1 | 200 µM M1P | ~20 | - | [10] |
| 20 | 6.9 ± 1.1 | 3 µM M16 | 5.9 ± 1.1 | 200 µM M1P | 23 76 (in the presence of 200 µM G16) | 40.0 ± 3.0 | this paper |
| Gene | Mutation | Accordance Among Classifiers | CADD Scaled Scores | Allele Frequency |
|---|---|---|---|---|
| ALG6 | p.Leu453Val | 10/11 | 23.6 | 0.012 |
| ALG3 | p.Val362Ile | 7/11 | 25.5 | 0.001 |
| MPDU1 | p.Ala229Thr | 3/11 | 20.9 | 0.154 |
| p.Gly225Ser | 4/11 | 24.1 | 0.010 | |
| ALG12 | p.Ile393Val | 7/11 | 23.4 | 0.112 |
| ALG8 | p.Arg268Gln | 11/11 | 35 | 0.014 |
| ALG2 | p.Pro56Leu | 10/11 | 24.2 | 0.001 |
| ALG1 | p.Thr64Asn | 7/11 | 24.7 | 0.003 |
| p.Ala3Asp | 3/11 | 22.6 | 0.001 | |
| ALG9 | p.Ser255Leu | 5/11 | 23.4 | 0.003 |
| RFT1 | p.Ala185Thr | 8/11 | 26.8 | 0.017 |
| p.Ser16Cys | 10/11 | 23.9 | 0.001 | |
| SRD5A3 | p.His309Asp | 7/11 | 15.91 | 0.004 |
| DDOST | p.Arg315Gln | 6/11 | 29.9 | 0.003 |
| p.Thr400Ile | 10/11 | 32 | 0.001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citro, V.; Cimmaruta, C.; Monticelli, M.; Riccio, G.; Hay Mele, B.; Cubellis, M.V.; Andreotti, G. The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers. Int. J. Mol. Sci. 2018, 19, 2218. https://doi.org/10.3390/ijms19082218
Citro V, Cimmaruta C, Monticelli M, Riccio G, Hay Mele B, Cubellis MV, Andreotti G. The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers. International Journal of Molecular Sciences. 2018; 19(8):2218. https://doi.org/10.3390/ijms19082218
Chicago/Turabian StyleCitro, Valentina, Chiara Cimmaruta, Maria Monticelli, Guglielmo Riccio, Bruno Hay Mele, Maria Vittoria Cubellis, and Giuseppina Andreotti. 2018. "The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers" International Journal of Molecular Sciences 19, no. 8: 2218. https://doi.org/10.3390/ijms19082218