Transcriptome Analysis of JA Signal Transduction, Transcription Factors, and Monoterpene Biosynthesis Pathway in Response to Methyl Jasmonate Elicitation in Mentha canadensis L.

Abstract
:1. Introduction
2. Results
2.1. Transcriptome Sequencing and De Novo Assembly of M. canadensis
2.2. Functional Annotation of Unigenes
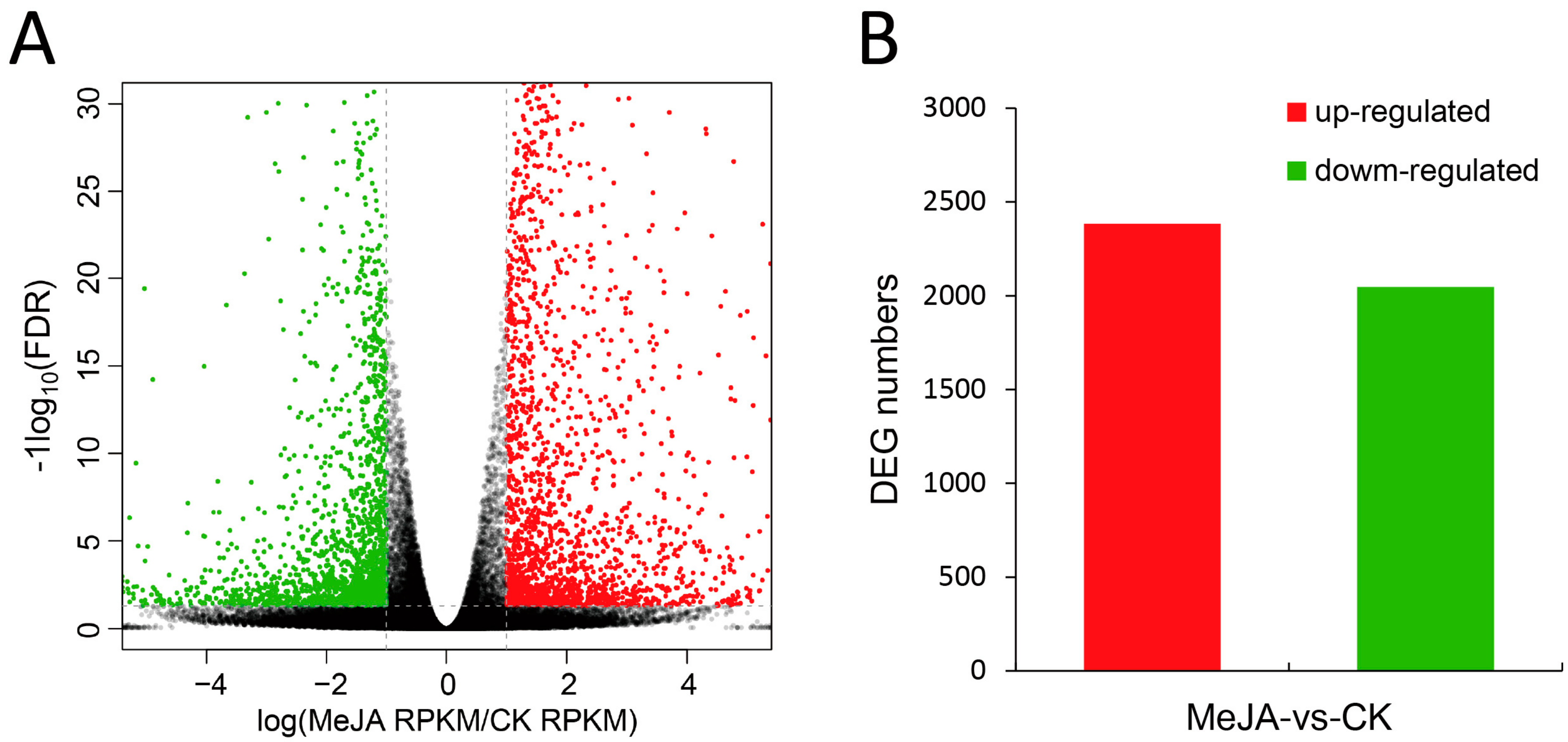
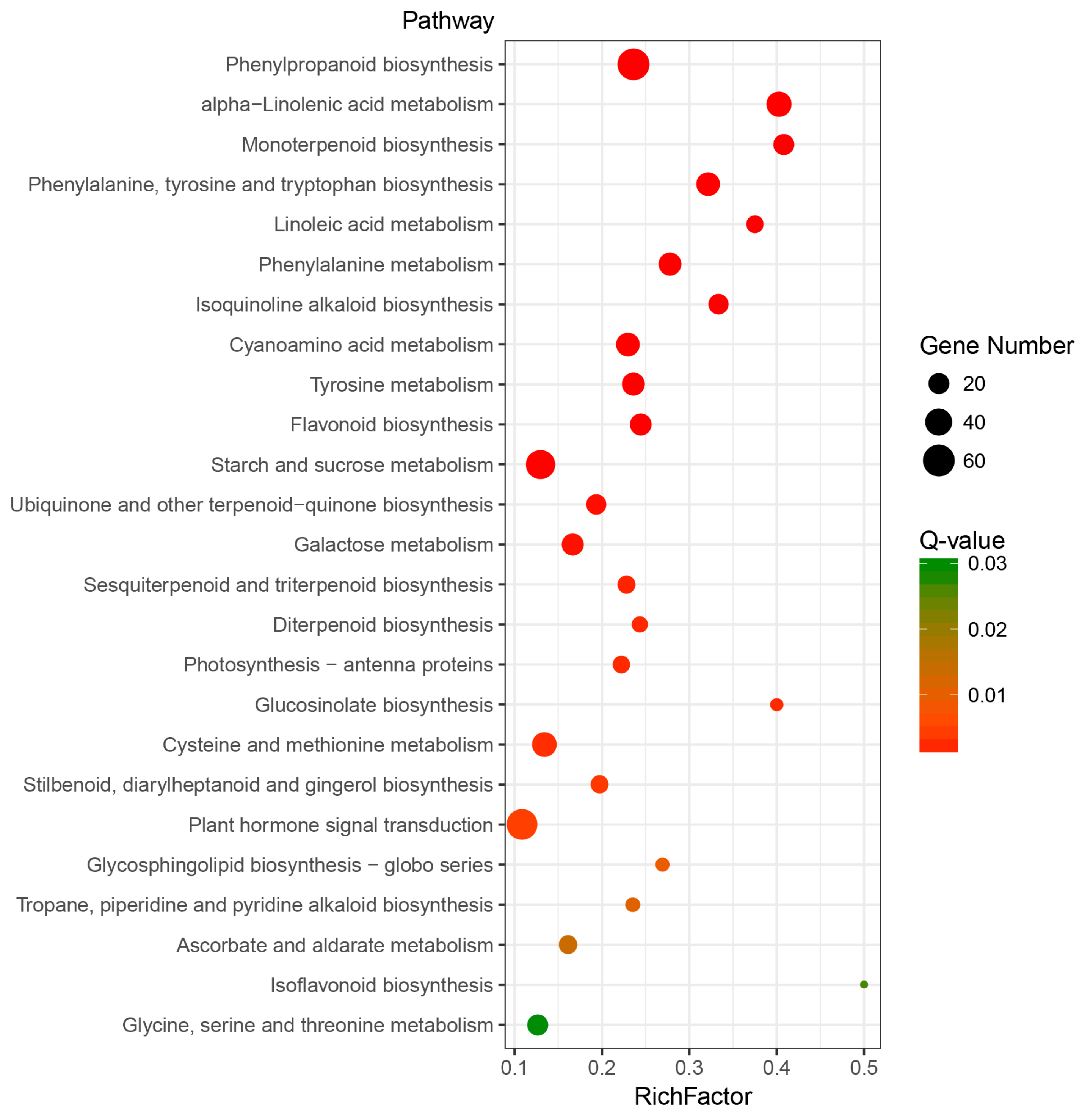
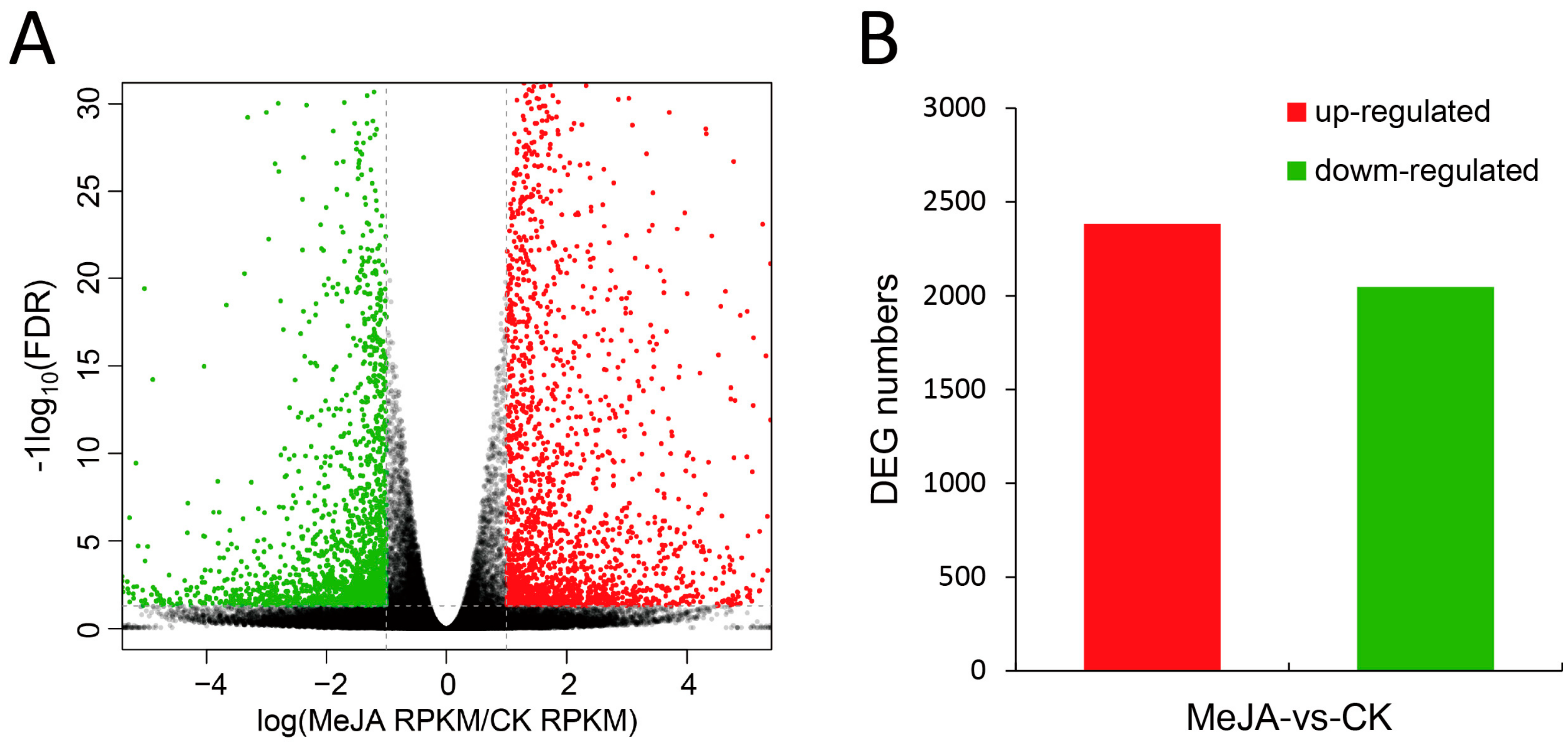
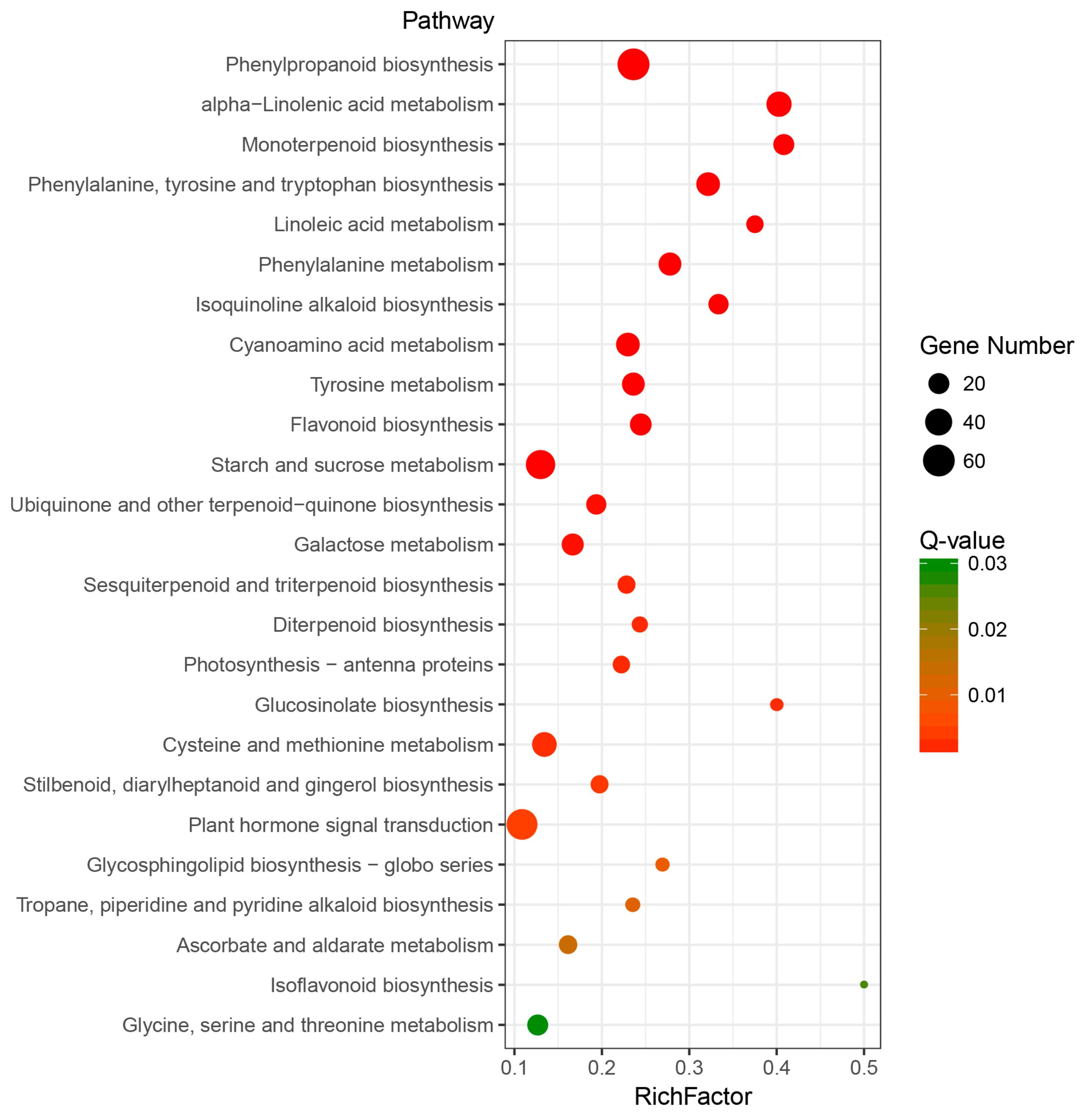
2.3. Identification and Analysis of Differentially Expressed Genes (DEGs)
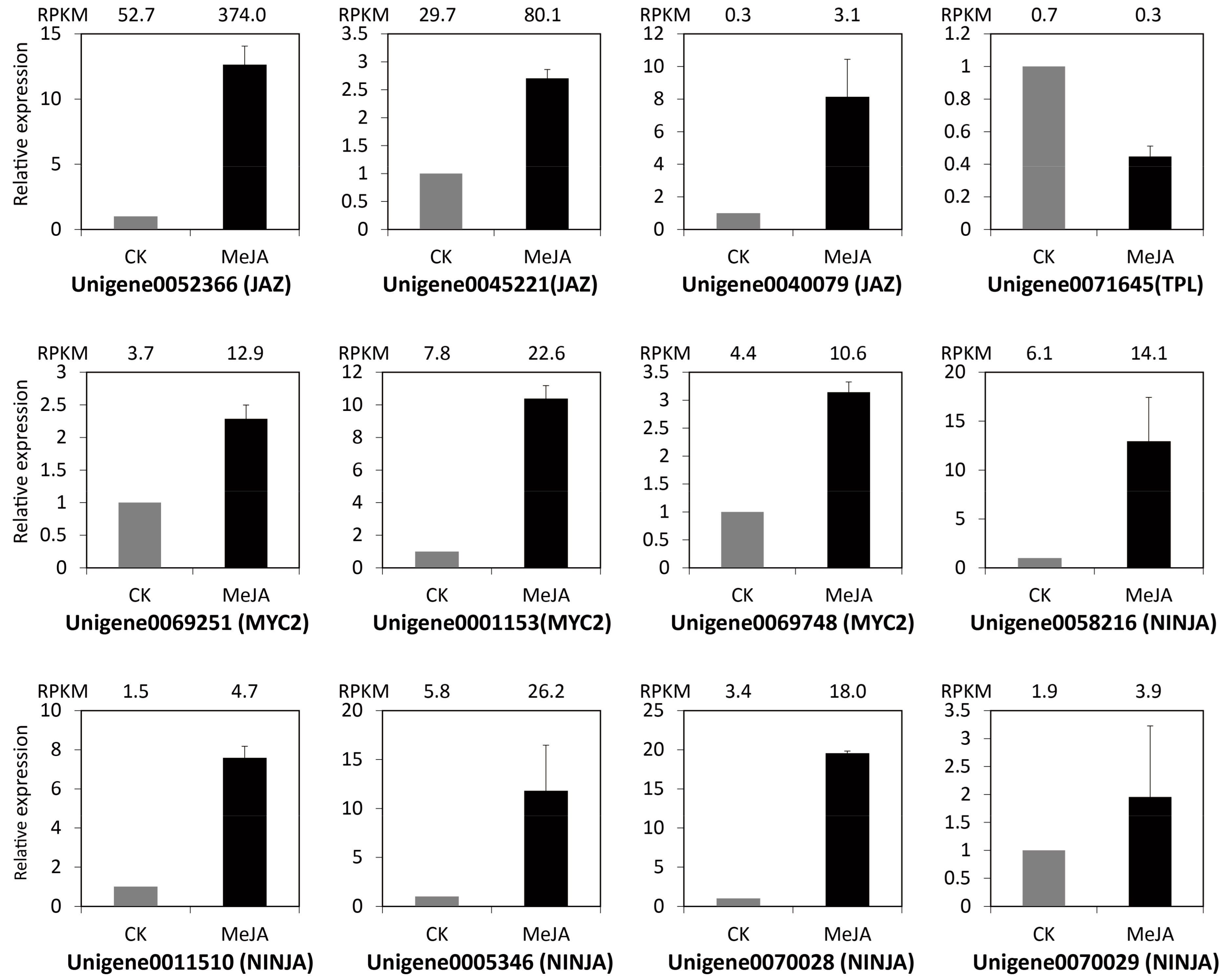
2.4. Identification and Expression Verification of JA Signal Pathway Genes under MeJA Treatment
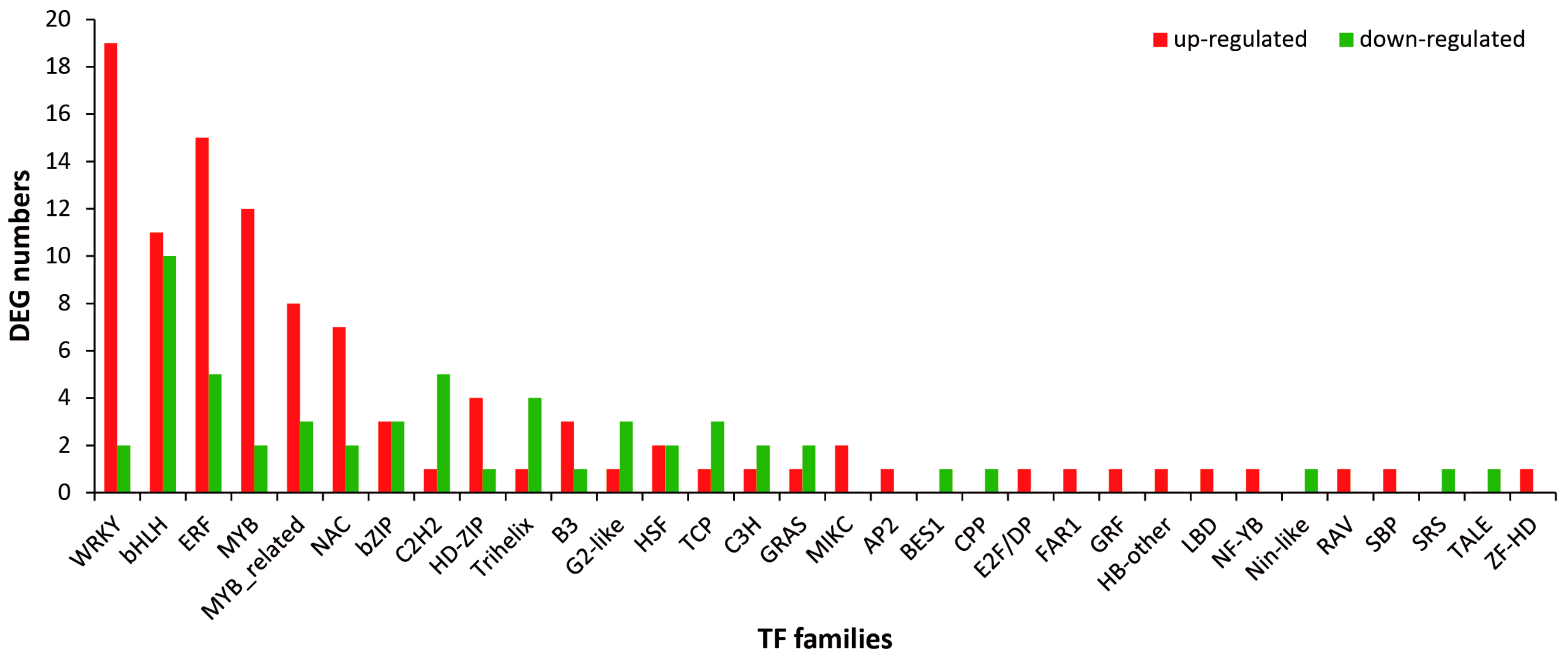
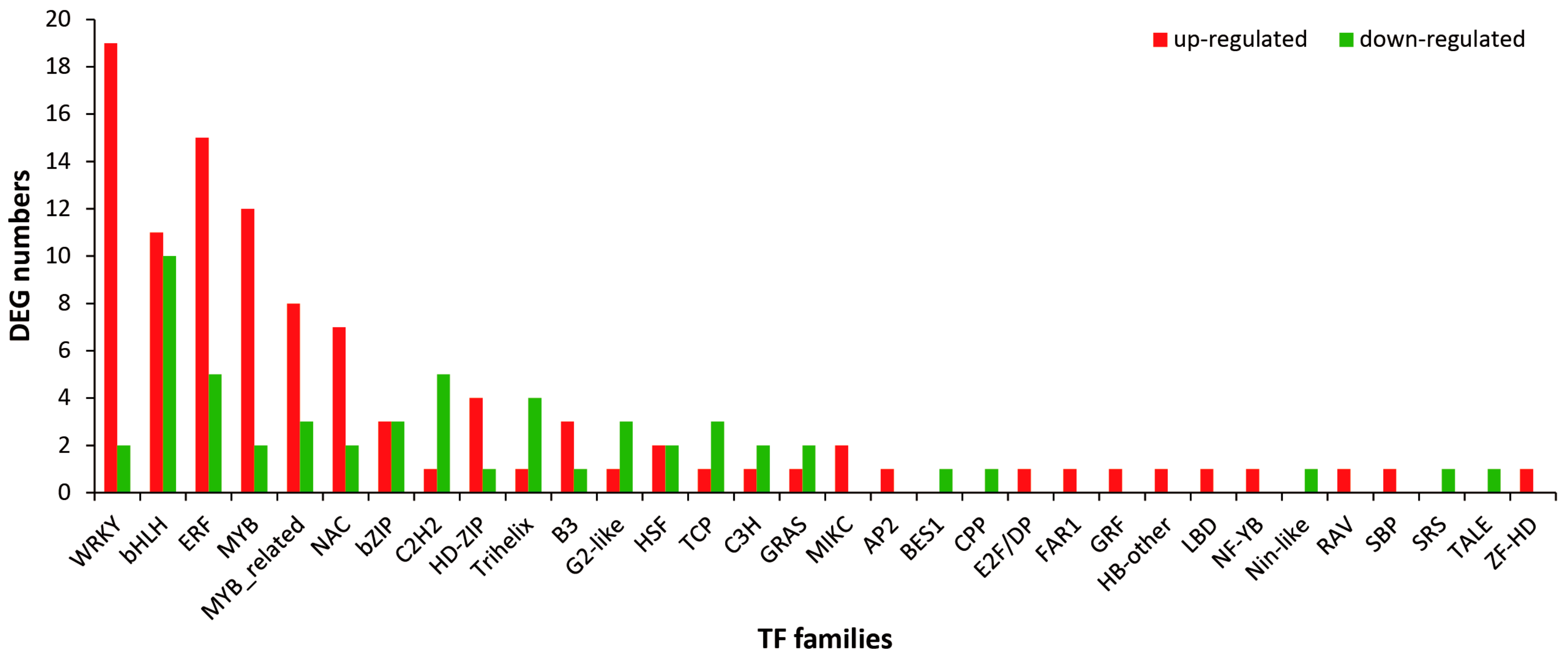
2.5. Identification and Analysis of Differentially Expressed TFs under MeJA Treatment
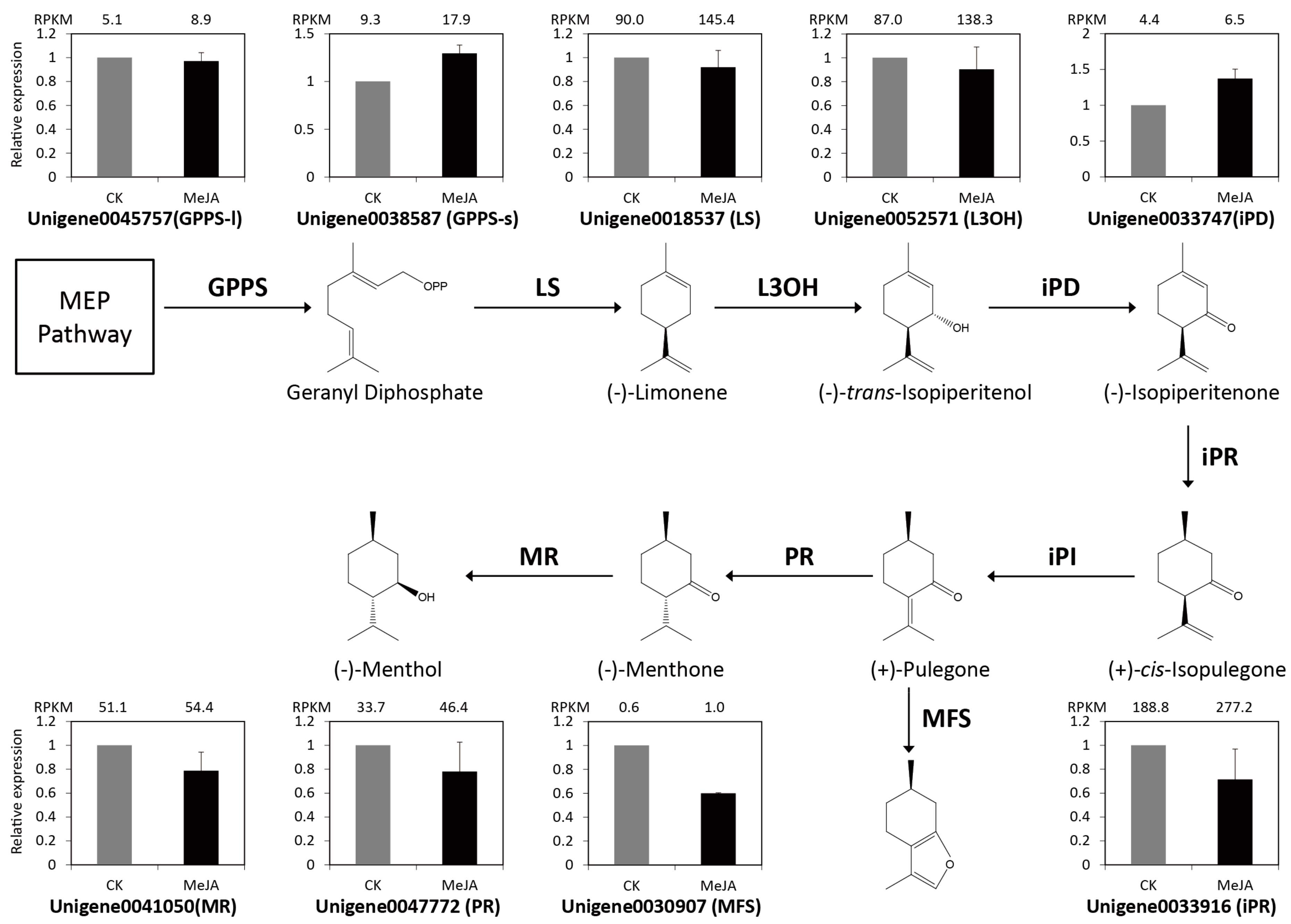
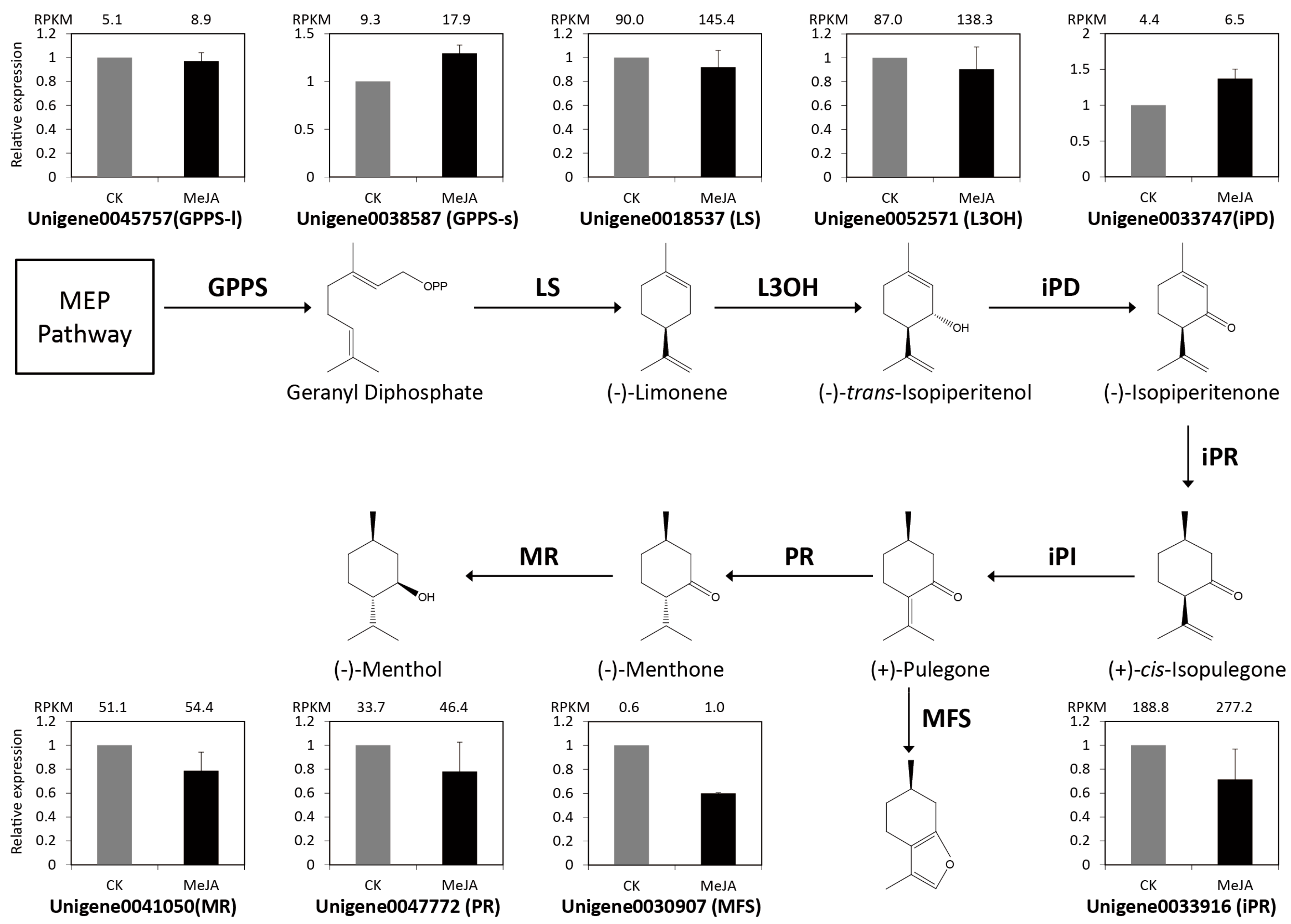
2.6. Identification and Expression Verification of Monoterpenoids and Menthol Biosynthetic Genes under MeJA Treatment
3. Discussion
4. Materials and Methods
4.1. Plant Material and JA Treatment
4.2. RNA Extraction, cDNA Library Construction and Illumina Sequencing
4.3. Transcriptome Assembly and Annotation
4.4. Differentially Expressed Genes Analysis
4.5. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MeJA | Methyl Jasmonate |
| CK | Control Check |
| MEP pathway | Methylerythritol Phosphate Pathway |
| GO | Gene Ontology |
| KOG | Eukaryotic Orthologous Group |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DEGs | Differentially Expressed Genes |
| RPKM | Reads per Kb per Million Reads |
| FDR | False Discovery Rate |
| qRT-PCR | Quantitative Real-Time PCR |
| TF | Transcription Factor |
References
- Lange, B.M.; Ahkami, A. Metabolic engineering of plant monoterpenes, sesquiterpenes and diterpenes-current status and future opportunities. Plant Biotechnol. J. 2013, 11, 169–196. [Google Scholar] [CrossRef] [PubMed]
- Croteau, R.; Karp, F.; Wagschal, K.C.; Satterwhite, D.M.; Hyatt, D.C.; Skotland, C.B. Biochemical characterization of a spearmint mutant that resembles peppermint in monoterpene content. Plant Physiol. 1991, 96, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.W.; Croteau, R. Organization of monoterpene biosynthesis in Mentha, immunocytochemical localizations of geranyl diphosphate synthase, limonene-6-hydroxylase, isopiperitenol dehydrogenase, and pulegone reductase. Plant Physiol. 2004, 136, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Croteau, R.B.; Davis, E.M.; Ringer, K.L.; Wildung, M.R. (−)-Menthol biosynthesis and molecular genetics. Naturwissenschaften 2005, 92, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Bertomeu, J.; Ros, R.; Arrillaga, I.; Segura, J. Expression of spearmint limonene synthase in transgenic spike lavender results in an altered monoterpene composition in developing leaves. Metab. Eng. 2008, 10, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Lange, B.M.; Mahmoud, S.S.; Wildung, M.R.; Turner, G.W.; Davis, E.M.; Lange, I.; Baker, R.C.; Boydston, R.A.; Croteau, R.B. Improving peppermint essential oil yield and composition by metabolic engineering. Proc. Natl. Acad. Sci. USA 2011, 108, 16944–16949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champagne, A.; Boutry, M. Proteomic snapshot of spearmint (Mentha spicata L.) leaf trichomes: A genuine terpenoid factory. Proteomics 2013, 13, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Lange, B.M. Biosynthesis and biotechnology of high-value p-menthane monoterpenes, including menthol, carvone, and limonene. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 319–353. [Google Scholar]
- Ahkami, A.; Johnson, S.R.; Srividya, N.; Lange, B.M. Multiple levels of regulation determine monoterpenoid essential oil compositional variation in the mint family. Mol. Plant 2015, 8, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopoeia Commission. The Pharmacopoeia of the People’s Republic of China, 2015 ed.; Medical Science Press: Beijing, China, 2015; 377p, ISBN 9787506744393. [Google Scholar]
- Zhao, D.; Xu, Y.W.; Yang, G.L.; Husaini, A.M.; Wu, W. Variation of essential oil of Mentha haplocalyx Briq. and Mentha spicata L. from China. Ind. Crops Prod. 2013, 42, 251–260. [Google Scholar] [CrossRef]
- Liang, C.Y.; Li, W.L.; Xia, B.; Liu, Y.; Yu, X. Chemical composition of essential oils of two Mentha species. Chem. Nat. Compd. 2010, 46, 656–657. [Google Scholar] [CrossRef]
- Cao, G.; Shan, Q.; Li, X.; Cong, X.; Zhang, Y.; Cai, H.; Cai, B. Analysis of fresh Mentha haplocalyx volatile components by comprehensive two-dimensional gas chromatography and high-resolution time-of-flight mass spectrometry. Analyst 2011, 136, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Ni, Y.; Kokot, S. A novel near-infrared spectroscopy and chemometrics method for rapid analysis of several chemical components and antioxidant activity of mint (Mentha haplocalyx Briq.) samples. Appl. Spectrosc. 2014, 68, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Yu, X.; Liu, Y.; Liang, C.Y.; Li, W.L. Analysis of genetic variability and relationships among Mentha L. using the limonene synthase gene, LS. Gene 2013, 524, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, L.; Inzé, D.; Goossens, A. Jasmonate-inducible gene: What does it mean? Trends Plant Sci. 2009, 14, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in annals of botany. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef] [PubMed]
- De Geyter, N.; Gholami, A.; Goormachtig, S.; Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012, 17, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Huang, J.J.; Luo, Z.Y. JA-mediated transcriptional regulation of secondary metabolism in medicinal plants. Sci. Bull. 2015, 60, 1062–1072. [Google Scholar] [CrossRef]
- Maes, L.; Van Nieuwerburgh, F.C.; Zhang, Y.; Reed, D.W.; Pollier, J.; Vande Casteele, S.R.; Inzé, D.; Covello, P.S.; Deforce, D.L.; Goossens, A. Dissection of the phytohormonal regulation of trichome formation and biosynthesis of the antimalarial compound artemisinin in Artemisia annua plants. New Phytol. 2011, 189, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Pu, G.; Lei, C.; Ma, L.; Wang, H.; Guo, Y.; Chen, J.; Du, Z.; Wang, H.; Li, G.; et al. Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 2009, 50, 2146–2161. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.X.; Li, J.X.; Yang, C.Q.; Hu, W.L.; Wang, L.J.; Chen, X.Y. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Jiang, W.; Zhang, L.; Zhang, F.; Zhang, F.; Shen, Q.; Wang, G.; Tang, K. AaERF1 positively regulates the resistance to Botrytis cinerea in Artemisia annua. PLoS ONE 2013, 8, e57657. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, S.; Aksoy, E. Genome-wide analysis of gene expression profiling revealed that COP9 signalosome is essential for correct expression of Fe homeostasis genes in Arabidopsis. Biometals 2017, 30, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gho, Y.S.; Jung, K.H.; Kim, S.R. Genome-wide identification and analysis of genes, conserved between japonica and indica rice cultivars, that respond to low-temperature stress at the vegetative growth stage. Front. Plant Sci. 2017, 8, 1120. [Google Scholar] [CrossRef] [PubMed]
- Di, F.; Jian, H.; Wang, T.; Chen, X.; Ding, Y.; Du, H.; Lu, K.; Li, J.; Liu, L. Genome-wide analysis of the PYL gene family and identification of PYL genes that respond to abiotic stress in Brassica napus. Genes 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zhong, Y.; Fu, X.; Lv, Z.; Shen, Q.; Yan, T.; Shi, P.; Ma, Y.; Chen, M.; Lv, X.; et al. Transcriptome analysis of genes associated with the artemisinin biosynthesis by jasmonic acid treatment under the light in Artemisia annua. Front. Plant Sci. 2017, 8, 971. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, T.; Wang, X.; Zhang, L.; Yang, M.; Chen, L.; Song, W.; Wang, C.; Chen, C. Transcriptome analyses from mutant Salvia miltiorrhiza reveals important roles for SmGASA4 during plant development. Int. J. Mol. Sci. 2018, 19, 2088. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yan, J.; Lei, J.; Li, J.; Zhu, J.; Zhang, H. De novo transcriptome sequencing of MeJA-induced Taraxacum koksaghyz Rodin to identify genes related to rubber formation. Sci. Rep. 2017, 7, 15697. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Guo, X.; Yang, X.; Wang, H.; Hua, W.; He, Y.; Kang, J.; Wang, Z. Transcriptional responses and gentiopicroside biosynthesis in methyl jasmonate-treated Gentiana macrophylla seedlings. PLoS ONE 2016, 11, e0166493. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Song, S. Jasmonates: Biosynthesis, metabolism, and signaling by proteins activating and repressing transciption. J. Exp. Bot. 2017, 68, 1303–1321. [Google Scholar] [CrossRef] [PubMed]
- Tholl, D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 2006, 9, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, L.; Morreel, K.; Witte, E.D.; Lammertyn, F.; Montagu, M.V.; Boerjan, W.; Inzé, D.; Goossens, A. Mapping methyl jasmonate-mediated transcriptional reprogramming of metabolism and cell cycle progression in cultured Arabidopsis cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Zhou, X.; Chen, X.; Li, Q.; Tan, H.; Dong, X.; Xiao, Y.; Chen, L.; Chen, W. Dynamic metabolic and transcriptomic profiling of methyl jasmonate-treated hairy roots reveals synthetic characters and regulators of lignan biosynthesis in Isatis indigotica Fort. Plant Biotechnol. J. 2016, 14, 2217–2227. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Nuruzzaman, M.; Xiu, H.; Huang, J.; Wu, K.; Chen, X.; Li, J.; Wang, L.; Jeong, J.H.; Park, S.J.; et al. Transcriptome analysis of methyl jasmonate-elicited Panax ginseng adventitious roots to discover putative ginsenoside biosynthesis and transport genes. Int. J. Mol. Sci. 2015, 16, 3035–3057. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 2016, 34, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C.; Pattanaik, S.; Patra, B.; Yuan, L. Analyses of Catharanthus roseus and Arabidopsis thaliana WRKY transcription factors reveal involvement in jasmonate signaling. BMC Genom. 2014, 15, 502. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, S.; Wang, N.; Xia, B.; Jiang, Y.; Wang, R. Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea. Front. Plant Sci. 2017, 7, 1971. [Google Scholar] [CrossRef] [PubMed]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, Y.; Xu, J.; Li, Y.; Luo, H.; Zhu, Y.; Liu, M.; Wu, Q.; Song, J.; Sun, C.; et al. Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from Panax quinquefolius. Plant Cell Tissue Organ Cult. 2013, 114, 269–277. [Google Scholar] [CrossRef]
- Suzuki, H.; Reddy, M.S.; Naoumkina, M.; Aziz, N.; May, G.D.; Huhman, D.V.; Sumner, L.W.; Blount, J.W.; Mendes, P.; Dixon, R.A. Methyl jasmonate and yeast elicitor induce differential transcriptional and metabolic re-programming in cell suspension cultures of the model legume Medicago truncatula. Planta 2005, 220, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fu, X.; Lv, Z.; Lu, X.; Shen, Q.; Zhang, L.; Zhu, M.; Wang, G.; Sun, X.; Liao, Z.; et al. A basic leucine zipper transcription factor, AabZIP1, connects abscisic acid signaling with artemisinin biosynthesis in Artemisia annua. Mol. Plant 2015, 8, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Lu, X.; Yan, T.; Fu, X.; Lv, Z.; Zhang, F.; Pan, Q.; Wang, G.; Sun, X.; Tang, K. The jasmonate-responsive AaMYC2 transcription factor positively regulates artemisinin biosynthesis in Artemisia annua. New Phytol. 2016, 210, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Wang, J.W.; Wang, S.; Wang, J.Y.; Chen, X.Y. Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-d-cadinene synthase-A. Plant Physiol. 2004, 135, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, J.; Mei, X. Enhancement of taxol production and excretion in Taxus chinensis cell culture by fungal elicitation and medium renewal. Appl. Microbiol. Biotechnol. 2001, 55, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Wang, L.F.; Dai, L.J.; Yang, S.G.; Tian, W.M. Characterization of HbEREBP1, a wound-responsive transcription factor gene in laticifers of Hevea brasiliensis Muell. Arg. Mol. Biol. Rep. 2012, 39, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wu, L.; Liang, J.; Shen, C.; Lin, J. Expression analysis and functional characterization of a novel cold-responsive gene CbCOR15a from Capsella bursa-pastoris. Mol. Biol. Rep. 2012, 39, 5169–5179. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Matsumoto, T.; Okada, A.; Komiyama, K.; Chujo, T.; Yoshikawa, H.; Nojiri, H.; Yamane, H.; Okada, K. Identification of target genes of the bZIP transcription factor OsTGAP1, whose overexpression causes elicitor-induced hyperaccumulation of diterpenoid phytoalexins in rice cells. PLoS ONE 2014, 9, e105823. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Reddy, V.A.; Panicker, D.; Mao, H.; Kumar, N.; Rajan, C.; Venkatesh, P.N.; Chua, N.; Sarojam, R. Metabolic engineering of terpene biosynthesis in plants using a trichome-specific transcription factor MsYABBY5 from spearmint (Mentha spicata). Plant Biotechnol. J. 2016, 14, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.A.; Wang, Q.; Dhar, N.; Kumar, N.; Venkatesh, P.N.; Rajan, C.; Panicker, D.; Sridhar, V.; Mao, H.Z.; Sarojam, R. Spearmint R2R3-MYB transcription factor MsMYB negatively regulates monoterpene production and suppresses the expression of geranyl diphosphate synthase large subunit (MsGPPS.LSU). Plant Biotechnol. J. 2017, 15, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A toolkit for illustrating heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples a | Raw Reads | Clean Reads | Reads Length (bp) | Clean Bases | Q20 Percentage (%) b | GC Percentage (%) |
|---|---|---|---|---|---|---|
| CK-1 | 58,914,374 | 57,759,958 (98.04%) | 150 | 8,536,291,577 | 98.69 | 50.79 |
| CK-2 | 58,863,024 | 57,904,740 (98.37%) | 150 | 8,588,205,461 | 98.88 | 50.05 |
| CK-3 | 58,109,454 | 57,113,908 (98.29%) | 150 | 8,470,135,931 | 98.83 | 50.69 |
| MeJA-1 | 61,076,212 | 59,885,296 (98.05%) | 150 | 8,855,787,485 | 98.69 | 50.55 |
| MeJA-2 | 55,398,094 | 54,424,124 (98.24%) | 150 | 8,072,356,234 | 98.82 | 50.46 |
| MeJA-3 | 65,765,946 | 64,630,794 (98.27%) | 150 | 9,590,111,719 | 98.82 | 50.39 |
| Total assembled bases | 59,279,270 |
| Unigene number | 81,843 |
| GC percentage (%) | 43.92 |
| N50 (bp) | 1126 |
| Average length (bp) | 724 |
| Annotation Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Nr | 52,700 | 64.39 |
| Swissprot | 34,565 | 42.23 |
| KOG | 29,536 | 36.09 |
| KEGG | 19,013 | 23.23 |
| Annotated in at least one database | 52,826 | 64.55 |
| Total unigenes | 81,843 | 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, X.; Fang, H.; Yu, X.; Xu, D.; Li, L.; Liang, C.; Lu, H.; Li, W.; Chen, Y.; Chen, Z. Transcriptome Analysis of JA Signal Transduction, Transcription Factors, and Monoterpene Biosynthesis Pathway in Response to Methyl Jasmonate Elicitation in Mentha canadensis L. Int. J. Mol. Sci. 2018, 19, 2364. https://doi.org/10.3390/ijms19082364
Qi X, Fang H, Yu X, Xu D, Li L, Liang C, Lu H, Li W, Chen Y, Chen Z. Transcriptome Analysis of JA Signal Transduction, Transcription Factors, and Monoterpene Biosynthesis Pathway in Response to Methyl Jasmonate Elicitation in Mentha canadensis L. International Journal of Molecular Sciences. 2018; 19(8):2364. https://doi.org/10.3390/ijms19082364
Chicago/Turabian StyleQi, Xiwu, Hailing Fang, Xu Yu, Dongbei Xu, Li Li, Chengyuan Liang, Hongfei Lu, Weilin Li, Yin Chen, and Zequn Chen. 2018. "Transcriptome Analysis of JA Signal Transduction, Transcription Factors, and Monoterpene Biosynthesis Pathway in Response to Methyl Jasmonate Elicitation in Mentha canadensis L." International Journal of Molecular Sciences 19, no. 8: 2364. https://doi.org/10.3390/ijms19082364