Selenium-Related Transcriptional Regulation of Gene Expression



Abstract
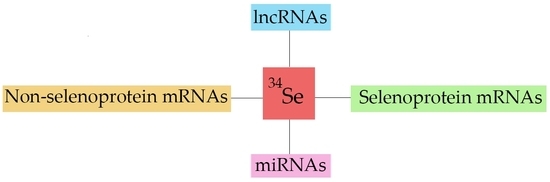
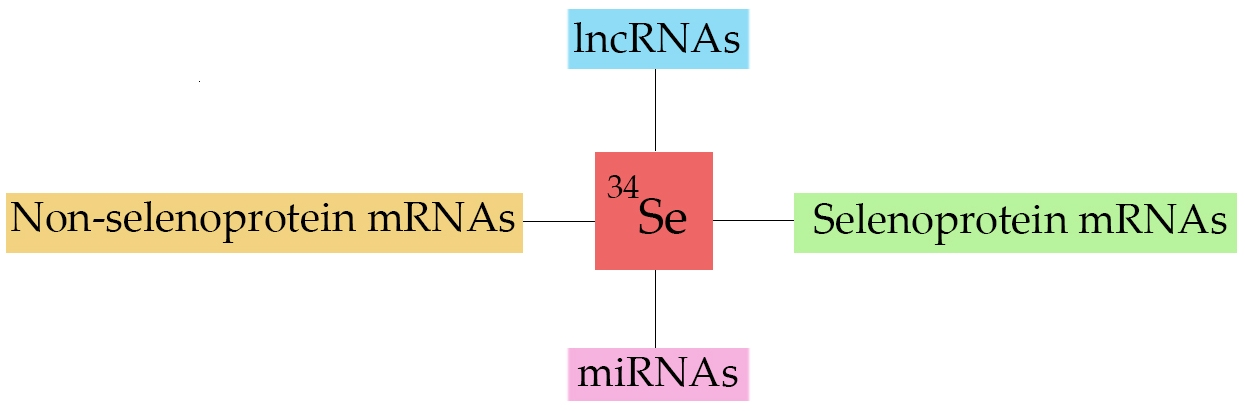
:
{kind=link}
1. Introduction
2. Selenoprotein Functions and Selenoprotein-Related Disorders
3. Biosynthesis of Selenocysteine and Selenoproteins
4. Regulation of Selenoprotein Transcriptome by Selenium
4.1. Glutathione Peroxidases
4.2. Thioredoxin Reductases
4.3. Iodothyronine Deiodinases
4.4. Other Selenoproteins
5. Regulation of Non-Selenoprotein Transcriptome by Selenium
6. Selenium Regulation of MicroRNAs and Long Non-Coding RNAs
7. Selenium-Related Gene Expressions in Some Pathological Conditions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| eIF4A3 | Translation initiation factor 4A3 |
| lncRNA | Long non-coding RNA |
| miRNA | Micro RNA |
| mRNA | Messenger RNA |
| NMD | Nonsense-mediated decay |
| Nrf2 | Nuclear factor, erythroid-2 like factor 2 |
| PPAR | Peroxisome proliferator-activated receptor |
| SBP2 | SECIS-binding protein 2 |
| Sec | Selenocysteine |
| SECIS | Selenocysteine insertion sequence |
| tRNA | Transfer RNA |
| tRNA[Ser] Sec | Selenocysteine transfer RNA |
| UTR | 3′-Untranslated region of messenger RNA |
| T3 | 3,5,3′-Triiodothyronine |
| T4 | 3,5,3,5″-Tetraiodothyronine |
References
- Kieliszek, M.; Blazejak, S. Current knowledge on the importance of selenium in food for living organisms: A review. Molecules 2016, 21, 609. [Google Scholar] [CrossRef] [PubMed]
- Whanger, P.D. Selenocompounds in plants and animals and their biological significance. J. Am. Coll. Nutr. 2002, 21, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hartikainen, H.L. Biochemistry of selenium and its impact on food chain quality and human health. J. Trace Elem. Med. Biol. 2005, 18, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Holben, D.H.; Smith, A.M. The diverse role of selenium within selenoproteins. J. Am. Diat. Assoc. 1999, 99, 836–843. [Google Scholar] [CrossRef]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: Synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Scientific opinion on dietary reference values for selenium. Eur. Food Saf. Auth. J. 2014, 12, 3846. [Google Scholar]
- Steinnes, E. Soils and geochemistry. Environ. Geochem. Health 2009, 31, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Kielczykowska, M.; Kocot, J.; Pazdzior, M.; Musik, I. Selenium—A fascinating antioxidant of protective properties. Adv. Clin. Exp. Med. 2018, 27, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Heseth, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Rathinasabapathi, B.; Wu, B.; Luo, J.; Pu, L.-P.; Ma, L.Q. Arsenic and selenium toxicity and their interactive effects in human. Environ. Int. 2014, 69, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Virtamo, J.; Valkeila, E.; Alftan, G.; Punsar, S.; Huttunen, J.K.; Karvonen, M.J. Serum selenium and the risk of coronary heart disease and stroke. Am. J. Epidemiol. 1985, 122, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhu, W.; Wang, W.; Li, R.; Hou, S.; Wang, D.; Yang, L. Selenium in soil and endemic diseases in China. Sci. Total Environ. 2002, 284, 227–235. [Google Scholar] [CrossRef]
- Han, J.; Guo, X.; Wang, L.; Chilufya, M.M.; Lim, P.N.; Qu, C. Selenium deficiency and selenium supplements: Biological effects on fibrosis in chronic diseases, from animal to human studies. In Handbook of Famine, Starvation, and Nutrient Deprivation; Preedy, V.R., Patel, V.B., Eds.; Springer: Cham, Switzerland, 2017; pp. 1–20. ISBN 987-3-319-40007-5. [Google Scholar]
- Steinbrenner, H.; Al-Quraishy, S.; Dkhil, M.A.; Wunderlich, F.; Sies, H. Dietary selenium in adjuvant therapy of viral and bacterial infections. Adv. Nutr. 2015, 6, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M.; Lipinski, B. Pathophysiological significance of protein hydrophobic interactions: An emerging hypothesis. Med. Hypotheses 2018, 110, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B. Sodium selenite as an anticancer agent. Anticancer Agents Med. Chem. 2017, 17, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Kieliszek, M.; Lipinski, B.; Blazejak, S. Application of sodium selenite in the prevention and treatment of cancers. Cells 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, B. Rationale for the treatment of cancer with sodium selenite. Med. Hypotheses 2005, 64, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Kiremidjian-Schumacher, L.; Roy, M.; Cohen, M.W.; Stotzky, G. Supplementation with selenium augments the functions of natural killer and lymphokine-activated killer cells. Biol. Trace Elem. 1996, 52, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and toxicity of sodium selenite in the treatment of patients with carcinoma in a phase I clinical trial: The SECAR study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.R.; Macey, M.G.; Lin, H.X.; Newland, A.C. The anti-leukaemic effects and the mechanism of sodium selenite. Leuk. Res. 1992, 16, 347–352. [Google Scholar] [CrossRef]
- Olm, E.; Jönsson-Videsäter, K.; Ribera-Cortada, I.; Fernandes, A.P.; Eriksson, L.C.; Lehmann, S.; Björnstedt, M. Selenite is a potent cytotoxic agent for human primary AML cells. Cancer Lett. 2009, 282, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, W.C.; Tappel, A.L. In vitro synthesis of glutathione peroxidase from selenite. Translational incorporation of selenocysteine. Biochim. Biophys. Acta 1983, 739, 225–234. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-dependent antioxidant enzymes: Actions and properties of selenoproteins. Antioxidants 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Bösl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5532–5534. [Google Scholar] [CrossRef]
- Kasaikina, M.V.; Hatfield, D.L.; Gladyshev, V.N. Understanding selenoprotein function and regulation through the use of rodent models. Biochim. Biophys. Acta 2012, 1823, 1633–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.A.; Hoffmann, P.R. The human selenoproteome: Recent insights into functions and regulation. Cell. Mol. Life Sci. 2009, 66, 2427–2478. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Hubert, N.; Walczak, R.; Sturchler, C.; Myslinski, E.; Schuster, C.; Westhof, E.; Carbon, P.; Krol, A. RNAs mediating cotranslational insertion of selenocysteine in eukaryotic selenoproteins. Biochimie 1996, 78, 590–596. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Orphan selenoproteins. BioEssays 1999, 21, 231–237. [Google Scholar] [CrossRef]
- Kim, H.Y.; Gladyshev, V.N. Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-R-sulfoxide reductases. PLoS Biol. 2005, 3, e375. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Fradejas-Villar, N. Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 2016, 30, 3669–3681. [Google Scholar] [CrossRef] [PubMed]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Quijano Roy, S.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat. Genet. 2001, 29, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Schweizer, U.; Holtmann, B.; Flohé, L.; Sendtner, M.; Köhrle, J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 2003, 370, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Zhou, J.; McMahan, W.J.; Motley, A.K.; Atkins, J.F.; Gesteland, R.F.; Burk, R.F. Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 2003, 278, 13640–13646. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Mears, A.J.; Bunker, R.; Ahmed, A.; MacKenzie, M.; Swarzentruber, J.A.; Beaulieu, C.L.; Ferretti, E.; FORGE Canada Consortium; Majewski, J.; et al. Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J. Med. Genet. 2014, 51, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 2004, 24, 9414–9423. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R; Chan, L.F.; Hughes, C.R.; Kaski, J.P.; Kowalczyk, J.C.; Savage, M.O.; Peters, C.J.; Nathwani, N.; Clark, A.J.L.; Storr, H.L.; et al. Thioredoxin reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J. Clin. Endocrinol. Metab. 2014, 99, E1556–E1563. [Google Scholar] [CrossRef] [PubMed]
- Bassett, J.H.D.; Boyde, A.; Howell, P.G.T.; Bassett, R.H.; Galliford, T.M.; Archanco, M.; Evans, H.; Lawson, M.A.; Croucher, P.; St. Germain, D.L.; et al. Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 7604–7609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, A.M.; Refetoff, S. The syndromes of reduced sensitivity to thyroid hormone. Biochim. Biophys. Acta 2013, 1830, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenmakers, E.; Carlson, B.; Agostini, M.; Moran, C.; Rajanayagam, O.; Bochukova, E.; Tobe, R.; Peat, R.; Gevers, E.; Muntoni, F.; et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Investig. 2016, 126, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, C.M.; Horton, C.R.; Carlson, B.A.; Parsons, T.E.; Hatfield, D.L.; Hallgrimsson, B.; Jirik, F.R. Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: A putative model for Kashin-Beck disease. PLoS Genet. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ma, W.J.; Zhang, F.; Ren, F.L.; Qu, C.J.; Lammi, M.J. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthritis Cartilage 2014, 22, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Chavatte, L. Update on selenoprotein biosynthesis. Antioxid. Redox Signal. 2015, 23, 775–794. [Google Scholar] [CrossRef] [PubMed]
- Mäenpää, P.H.; Bernfield, M.R. A specific hepatic transfer RNA for phosphoserine. Proc. Natl. Acad. Sci. USA 1970, 67, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.; Portugal, F.H. Seryl-tRNA in mammalian tissues: Chromatographic differences in brain and liver and a specific response to the codon, UGA. Proc. Natl. Acad. Sci. USA 1970, 67, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Low, S.C.; Harney, J.W.; Berry, M.J. Cloning and functional characterization of human selenophosphate synthetase, an essential component of selenoprotein synthesis. J. Biol. Chem. 1995, 270, 21659–21664. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-M.; Carlson, B.A.; Irons, R.; Mix, H.; Zhong, N.; Gladyshev, V.N. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem. J. 2007, 404, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Latreche, L.; Jean-Jean, O.; Driscoll, D.M.; Chavatte, L. Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009, 37, 5868–5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.W., 3rd; Harney, J.W.; Berry, M.J. Selenecysteine incorporation in eukaryotes: Insights into mechanism and efficiency from sequence, structure, and spacing proximity studies of the type I deiodinase SECIS element. RNA 1996, 2, 171–182. [Google Scholar] [PubMed]
- Copeland, P.R.; Fletcher, J.E.; Carlson, B.A.; Hatfield, D.L.; Driscoll, D.M. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 2000, 19, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, S.L.; Grundner-Culemann, E.; Harney, J.W.; Berry, M.J. SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J. 2000, 19, 6882–6890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latreche, L.; Duhieu, S.; Touat-Hamici, Z.; Jean-Jean, O.; Chavatte, L. The differential expression of glutathione peroxidase 1 and 4 depends on the nature of SECIS element. RNA Biol. 2012, 9, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Stoytchev, I.; Forry, E.P.; Berry, M.J. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol. Cell. Biol. 2007, 27, 7848–7855. [Google Scholar] [CrossRef] [PubMed]
- Kervestin, S.; Jacobson, A. NMD: A multifaceted response to premature translational termination. Nat. Rev. Mol. Cell. Biol. 2012, 13, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Budiman, M.E.; Bubenik, J.L.; Miniard, A.C.; Middleton, L.M.; Gerber, C.A.; Cash, A.; Driscoll, D.M. Eukaryotic initiation factor 4a3 is a selenium-regulated RNA-binding protein that selectively inhibits selenocysteine incorporation. Mol. Cell 2009, 35, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Seyedali, A.; Berry, M.J. Nonsense-mediated decay factors are involved in the regulation of selenoprotein mRNA levels during selenium deficiency. RNA 2014, 20, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavatte, L.; Brown, B.A.; Driscoll, D.M. Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 2005, 12, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Shen, Q.; Newburger, P.E. Recognition and binding of the human selenocysteine insertion sequence by nucleolin. J. Cell. Biochem. 2000, 77, 507–516. [Google Scholar] [CrossRef]
- Lei, X.G.; Evenson, J.K.; Thompson, K.M.; Sunde, R.A. Glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase are differently regulated in rats by dietary selenium. J. Nutr. 1995, 125, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Sunde, R.A.; Raines, A.M.; Barnes, K.M.; Evenson, J.K. Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 2009, 29, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.P.; Fu, J.; Lin, S.L; Wang, X.S.; Li, S. Effect of dietary selenium deficiency on mRNA levels of twenty-one selenoprotein genes in the liver of layer chicken. Biol. Trace Elem. Res. 2014, 159, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Bermano, G.; Nicol, F.; Dyer, J.A.; Sunde, R.A.; Beckett, G.J.; Arthur, J.R.; Hesketh, J.E. Tissue-specific regulation of selenoenzyme gene expression in rats. Biochem. J. 1995, 311, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. 2008, 72, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione metabolism. Methods Enzymol. 1995, 251, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. The incomplete glutathione puzzle: Just guessing at numbers and figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. 2017, 131, 1669–1688. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, S.; Asano, Y.; Arai, K.; Iwaoka, M. Selenogluthione diselenide: Unique redox reactions in the GPx-like catalytic cycle and repairing of disulfide bonds in scrambled protein. Biochemistry 2017, 56, 5644–5653. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kumakura, F.; Komatsu, I.; Arai, K.; Onuma, Y.; Hojo, H.; Singh, B.G.; Priyadarsini, K.I.; Iwaoka, M. Antioxidative glutathione peroxidase activity of selenoglutathione. Angew. Chem. Int. Ed. Engl. 2011, 50, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, A.; Rigobello, M.P. Principles in redox signaling; from chemistry to functional significance. Antioxid. Redox Signal. 2013, 18, 1557–1593. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [PubMed]
- Chu, F.-F.; Esworthy, S.R. The expression of an intestinal form of glutathione peroxidase (GSHPx-GI) in rat intestinal epithelium. Arch. Biochem. Biophys. 1995, 323, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.-F.; Doroshow, J.H.; Esworthy, R.S. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J. Biol. Chem. 1993, 268, 2571–2576. [Google Scholar] [PubMed]
- Wingler, K.; Böcher, M.; Flohe, L.; Kollmus, H.; Brigeliu-Flohe, R. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. FEBS J. 1999, 259, 149–157. [Google Scholar] [CrossRef]
- Avissar, N.; Ornt, D.B.; Yagil, Y.; Horowitz, S.; Watkins, R.H.; Kerl, E.A.; Takahashi, K.; Palmer, I.S.; Cohen, H.J. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am. J. Physiol. 1994, 266, C367–C375. [Google Scholar] [CrossRef] [PubMed]
- Olson, G.E.; Whitin, J.C.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Austin, L.M.; Deal, J.; Cohen, H.J.; Burk, R.F. Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. 2010, 298, F1244–F1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohe, R.; Aumann, K.D.; Blocker, H.; Gross, G.; Kiess, M.; Kloppel, K.D.; Maiorino, M.; Roveri, A.; Schuckelt, R.; Ursini, F.; et al. Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA, and deduced amino acid sequence. J. Biol. Chem. 1994, 269, 7342–7348. [Google Scholar] [PubMed]
- McCann, J.C.; Ames, B.N. Adaptive dysfunction of selenoproteins from the perspective of the triage theory: Why modest selenium deficiency may increase risk of diseases of aging. FASEB J. 2011, 25, 1793–1814. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-A.; Wu, Y.; Zappacosta, F.; Jeang, K.-T.; Lee, B.J.; Hatfield, D.L.; Gladyshov, V.N. Redox regulation of cell signaling by selenocysteine in mammalian thioredoxin reductases. J. Biol. Chem. 1999, 274, 24522–24530. [Google Scholar] [CrossRef] [PubMed]
- Lothrop, A.P.; Ruggles, E.L.; Hondal, R.J. No selenium required: Reactions catalyzed by mammalian thioredoxin reductase that are independent of selenocysteine residue. Biochemistry 2009, 48, 6213–6223. [Google Scholar] [CrossRef] [PubMed]
- Lothrop, A.P.; Snider, G.W.; Ruggles, E.L; Hondal, R.J. Why is mammalian reductase 1 so dependent upon the use of selenium? Biochemistry 2014, 53, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Venardos, K.; Ashton, K.; Headrick, J.; Perkins, A. Effects of dietary selenium on post-ischemic expression of antioxidant mRNA. Mol. Cell. Biochem. 2005, 270, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.N.; Cassidy, A.; Wang, W.; Wei, T.; Belshaw, N.J; Johnson, I.T.; Brigelius-Flohe, R.; Bao, Y. TrxR1 and Gpx2 are potently induced by isothiocyanates and selenium, and mutually cooperate to protect Caco-2 cells against free radical-mediated cell death. Biochim. Biophys. Acta 2012, 1823, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Legrain, Y.; Touat-Hamici, Z.; Chavatte, L. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fiubroblasts. J. Biol. Chem. 2014, 289, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.J. Targeting the selenoprotein thioredoxin reductase 1 for anticancer therapy. Adv. Cancer Res. 2017, 136, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Rundlöf, A.-K.; Arnér, E.S.J. Regulation of the mammalian selenoprotein thiredoxin reductase 1 in relation to cellular phenotype, growth, and signaling events. Antioxid. Redox Signal. 2004, 6, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Rundlöf, A.-K.; Janard, M.; Miranda-Vizuete, A.; Arnér, E.S.J. Evidence for intriguingly complex transcription of human thioredoxin reductase 1. Free Radic. Biol. Med. 2004, 36, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Björkhem-Bergman, L.; Jönsson, K.; Eriksson, L.C.; Olsson, J.M.; Lehmann, S.; Paul, C.; Björnstedt, M. Drug-resistant human lung cancer cells are more sensitive to selenium cytotoxicity. Effects on thioredoxin reductase and glutathione reductase. Biochem. Pharmacol. 2002, 63, 1875–1884. [Google Scholar] [CrossRef]
- Hernandez, A.; St. Germain, D.L. Thyroid hormone deiodinases: Physiology and clinical disorders. Curr. Opin. Pediatr. 2003, 15, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Beckett, G.J; MacDougall, D.A.; Nicol, F.; Arthur, R. Inhibition of type I and type II iodothyronine deiodinase activity in rat liver, kidney and brain produced by selenium deficiency. Biochem. J. 1989, 259, 887–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behne, D.; Kyriakopoulos, A.; Meinhold, H.; Köhrle, J. Identification of type I iodothyronine 5’-deiodinase as a selenoenzyme. Biochem. Biophys. Res. Commun. 1990, 173, 1143–1149. [Google Scholar] [CrossRef]
- Hill, K.E.; Lyons, P.R.; Burk, R.F. Differential regulation of rat liver selenoprotein mRNAs in selenium deficiency. Biochem. Biophys. Res. Commun. 1992, 185, 260–263. [Google Scholar] [CrossRef]
- Koenig, R.J. Regulation of type I iodothyronine deiodinase in health and disease. Thyroid 2005, 15, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Foroughi, M.A.; Dehghani, H. Short communication: Quantitative comparison of iodothyronine deiodinase I and II mRNA expression in ovine tissues. Res. Vet. Sci. 2013, 95, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 2005, 25, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, M.J.; Peterson, D.; Vicari, A.; Cocks, B.G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Ferrick, D.A.; Kastelein, R.A.; Bazan, J.F.; et al. Identification of a novel selD homolog from eukaryotes, bacteria, and archaea: Is there an autoregulatory mechanism in selenocysteine metabolism? Proc. Natl. Acad. Sci. USA 1996, 93, 15086–15091. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; Höge, S.C.; Hoffmann, F.W.; Hashimoto, A.C.; Barry, M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007, 35, 3963–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raines, A.M.; Sunde, R.A. Selenium toxicity but not deficient or supernutrional selenium status vastly alters the transcriptome in rodents. BMC Genom. 2011, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zheng, Y.; Min, Z.; Ning, Q.; Lu, S. Selenium effect on selenoprotein trasncriptome in chondrocytes. Biometals 2013, 26, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Sunde, R.A.; Raines, A.M. Selenium regulation of the selenoprotein and nonselenoprotein transcriptomes in rodents. Adv. Nutr. 2011, 2, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Pohjanvirta, R.; Boutros, P.C.; Moffat, I.D.; Lindén, J.; Wendelin, D.; Okey, A.B. Genome-wide effects of acute progressive feed restriction in liver and white adipose tissue. Toxicol. Appl. Pharmacol. 2008, 230, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Dominguez, A.; Swan, D.; Ford, D.; Hesketh, J. Selenium alters miRNA profile in an intestinal cell line. Evidence that miR-185 regulates expression of GPX2 and SEPSH2. Mol. Nutr. Food Res. 2013, 57, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Matouskova, P.; Hanouskova, B.; Skalova, L. MicroRNAs as potential regulators of glutathione peroxidases expression and their role in obesity and related pathologies. Int J. Mol. Sci. 2018, 19, 1199. [Google Scholar] [CrossRef] [PubMed]
- Potenza, N.; Castiello, F.; Panella, M.; Colonna, G.; Ciliberto, G.; Russo, A.; Costantini, S. Human miR.544a modulates SELK expression in hepatocarcinoma cell lines. PLoS ONE 2016, 11, e0156908. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gu, D.; Zhou, M.; Shi, H.; Yan, S.; Cai, Y. Regulatory role of miR-125a/b in the suppression by selenium of cadmium-induced apoptosis via the mitochondrial pathway in LLC-PK1 cells. Chem. Biol. Interact. 2016, 243, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Liu, Z.; Yang, G.; Gao, D.; Niu, X. MicroRNA expression profiles in rats with selenium deficiency and the possible role of the Wnt/β-catenin signaling pathway in cardiac dysfunction. Int. J. Mol. Med. 2015, 35, 143–152. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.J.; Rotjanapun, K.; Hesketh, J.E.; Roy, N.C. Expression profiling indicating low selenium-sensitive microRNA levels linked to cell cycle and stress response pathways in the Caco-2 cell line. Br. J. Nutr. 2017, 117, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Fan, R.; Zhao, J.; Zhao, X.; Yang, J.; Zhang, Z.; Xu, S. Impact of exudative diathesis induced by selenium deficiency on lncRNAs and their roles in the oxidative reduction process in broiler chick veins. Oncotarget 2017, 8, 20695–20705. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Cao, C.; Zhao, X.; Shi, Q.; Zhao, J.; Xu, S. Downregulated long noncoding RNA ALDBGAL0000005049 induces inflammation in chicken muscle suffered from selenium deficiency by regulating stearoyl-CoA desaturase. Oncotarget 2017, 8, 52761–52774. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lv, Y.; Wang, Y.; Du, P.; Tan, W.; Lammi, M.J.; Guo, X. Network analysis of Se- and Zn-related proteins in the serum proteomics expression profile of the endemic dilated cardiomyopathy Keshan disease. Biol. Trace Elem. Res. 2018, 183, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yan, R.; Wang, B.; Du, P.; Tan, W.; Lammi, M.J.; Guo, X. Prediction of co-expression genes and integrative analysis of gene microarray proteomics profile of Keshan disease. Sci. Rep. 2018, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Z.; Guo, X.; Duan, C.; Ma, W.J.; Zhang, Y.G.; Xu, P.; Gao, Z.Q.; Wang, Z.F.; Yan, H.; Zhang, Y.F.; et al. Comparative analysis of gene expression profiles between the normal human cartilage and the one with endemic osteoarthritis. Osteoarthritis Cartilage 2009, 17, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Guo, X.; Zhang, X.D.; Yu, H.J.; Yan, H.; Gao, Y.; Ma, W.J.; Gao, Z.Q.; Xu, P.; Lammi, M. Comparative analysis of gene expression profiles between primary knee osteoarthritis and an osteoarthritis endemic to Northwestern China, Kashin-Beck disease. Arthritis Rheum. 2010, 62, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Guo, X.; Duan, C.; Wu, S.; Yu, H.; Lammi, M. Identification of differentially expressed genes and pathways between primary osteoarthritis and endemic osteoarthritis (Kashin-Beck disease). Scand. J. Rheumatol. 2013, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, W.; Guo, X.; Zhang, F.; Ma, W.; Zhang, Y.; Li, Y.; Bai, Y.; Lammi, M.J. Pathways related to mitochondrial dysfunction in cartilage of endemic osteoarthritis patients in China. Sci. China Life Sci. 2012, 55, 1057–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Liu, H.; Zhang, F.; Shao, W.; Yang, L.; Ning, Y.; Wang, S.; Zhao, G.; Lee, B.J.; Lammi, M.; et al. Long noncoding RNA expression profile reveals lncRNAs signature associated with extracellular matrix degradation in kashin-beck disease. Sci. Rep. 2017, 7, 17553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Zhou, J.; Zhou, J. Selenium status of children in Kashin-Beck disease endemic areas in Shaanxi, China: Assessment with mercury. Environ. Geochem. Health 2018, 40, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Wang, X.; Zhang, P.; Anatoly, S.V.; Prakash, N.T.; Li, C.; Zhou, R.; Lammi, M.; Zhang, F.; Guo, X. Imbalance of dietary nutrients and the associated differentially expressed genes and pathways may play important roles in juvenile Kashin-Beck disease. J. Trace Elem. Med. Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lammi, M.J.; Qu, C. Selenium-Related Transcriptional Regulation of Gene Expression. Int. J. Mol. Sci. 2018, 19, 2665. https://doi.org/10.3390/ijms19092665
Lammi MJ, Qu C. Selenium-Related Transcriptional Regulation of Gene Expression. International Journal of Molecular Sciences. 2018; 19(9):2665. https://doi.org/10.3390/ijms19092665
Chicago/Turabian StyleLammi, Mikko J., and Chengjuan Qu. 2018. "Selenium-Related Transcriptional Regulation of Gene Expression" International Journal of Molecular Sciences 19, no. 9: 2665. https://doi.org/10.3390/ijms19092665
APA StyleLammi, M. J., & Qu, C. (2018). Selenium-Related Transcriptional Regulation of Gene Expression. International Journal of Molecular Sciences, 19(9), 2665. https://doi.org/10.3390/ijms19092665