1. Introduction
The discovery of antitumor activity of cis-platin (cis-PtCl
2(NH
3)
2) has led the world wide researches to investigate the antitumor activity of thousands compounds of platinum and other metals [
1,
2,
3,
4,
5]. The standard cis-diamminedichloroplatinum(II) has recently been the subject of great attention because of its well established antitumor activity [
1,
2,
3,
4,
5].
It is, however, expected that the new tumor-inhibiting metal compounds may also act against tumor other than those affected by cis-platin [
6,
7]. To the very promising antitumor agents of the third generation belong organometallic tin compounds [
4,
8]. Recently, a number of tin complexes were studied and in many cases they seem to manifest efficient anticancer activity [
4,
8,
9]. The advantage of their use as antitumor medicines lies in their lower costs and possibly in a gretaer effectivness. However, in general, a satisfactory understanding of the mode of action of the drug requires a thorugh study of its interaction with the relevant biomolecules or their model systems.
Based on quantum chemical calculations, the possible molecular structures of anticancerogenic platinum and organotin commpounds were considered. The molecular orbital calculations were employed in searching electronic parameters which may characterize the anticancerogenic action [
10]. It is well established that cis-platinum complexes interact with nucleosides blocks preferentially through the N7 site of guanine [
1,
2,
3]. It may also form bidentate chelates as a result of interaction of cis-platinum both with N7 and O6 sites [
11]. On the other hand the hydrogen bonding in Pt ammine complexes may stabilize the binding of Pt ammine cations to oligonucleotides [
12]. It seems therefore that the similar effects might be expected in the case of tin complexes.
2. Computational
The molecular and electronic structure of the systems under consideration have been studied both at the semiempirical level and with non-empirical ab initio methods. In the first case the PM3 of MOPAC2000 package was used [
13] and in the second one the quantum chemical calculations have been performed by using GAUSSIAN-98 computer program [
14]. All non-empirical calculations were performed within the Huzinaga basis set [
15] including the two-membered polarization functions with full geometry optimization at the MP2 level. On the contrary to the most attainable calculations, no restrictions on the symmetry were imposed in our case performing them in all cases for the C
1 point group. Despite of the fact that the computation of the all electron calculation is enormously time consuming, it seems to be most justified from the first quantum mechanical principles as well as most accurate. In our case the structural parameters obtained for the Pt(NH
3)
2Cl
2 complex [
16] are in the reasonable agreement with the relevant experimental results (
Table 1) [
17] and a slightly better than some other calculations [
18]. It should be noted that in the case of the lowest molecular symmetries the results of calaculation are most close to the experimental ones.
Table 1.
Ab initio calculated and experimental [
17] Pt-Cl and Pt-N bond lengths [Å] for cis and trans platin
Table 1.
Ab initio calculated and experimental [17] Pt-Cl and Pt-N bond lengths [Å] for cis and trans platin
| Symmetry | cis-Pt(NH3)2Cl2 | trans-Pt(NH3)Cl2 |
| Pt-Cl | Pt-N | Pt-Cl | Pt-N |
| C1 | 2.331 | 2.085 | 2.302 | 2.047 |
| C2v | 2.328 | 2.089 | 2.349 | 2.048 |
| | | | | |
| Exp. | 2.33 ± 0.01 | 2.02 ± 0.04 | 2.32 ± 0.01 | 2.05 ± 0.04 |
There are a number of earlier computational results concerning platinum anticancer compounds [
18,
19,
20,
21], and very recent one [
22]. On the contrary, however, quantum chemical calculations on tin containing anticancer compounds are rather scarce. They concern mainly semiempirical methods, as timely less expensive. These methods, as well as the LANL2DZ pseudopotential approach, were used for electronic structure investigations of dimethyltin(IV) and DNA model systems [
23,
24], dimethyl and trimethyl-tin(IV) complexes of porphyrin derivatives [
24].
3. Results and Conclusions
The fully optimized molecular structures i.e. optimal conformations of platinum and tin chelates are shown in
Fig 1,
Fig 2,
Fig 3,
Fig 4 and
Fig 5. The evaluated PM3 (with our preliminary parametrization for the Pt atom: USS=45.550, Upp=65.894, UDD=79.000, BETAS=-2.780, BETAP=-2.006, ZS=2.000, ZP=2.000, ZD=5.884, ALP=1.690, GSS=10.190, GSP=7.235, GPP=2.100, GP2=1.000, HSP=0.503 according to the package scheme [
13]) and ab initio binding energies of complexes under considerations are listed in
Table 2.
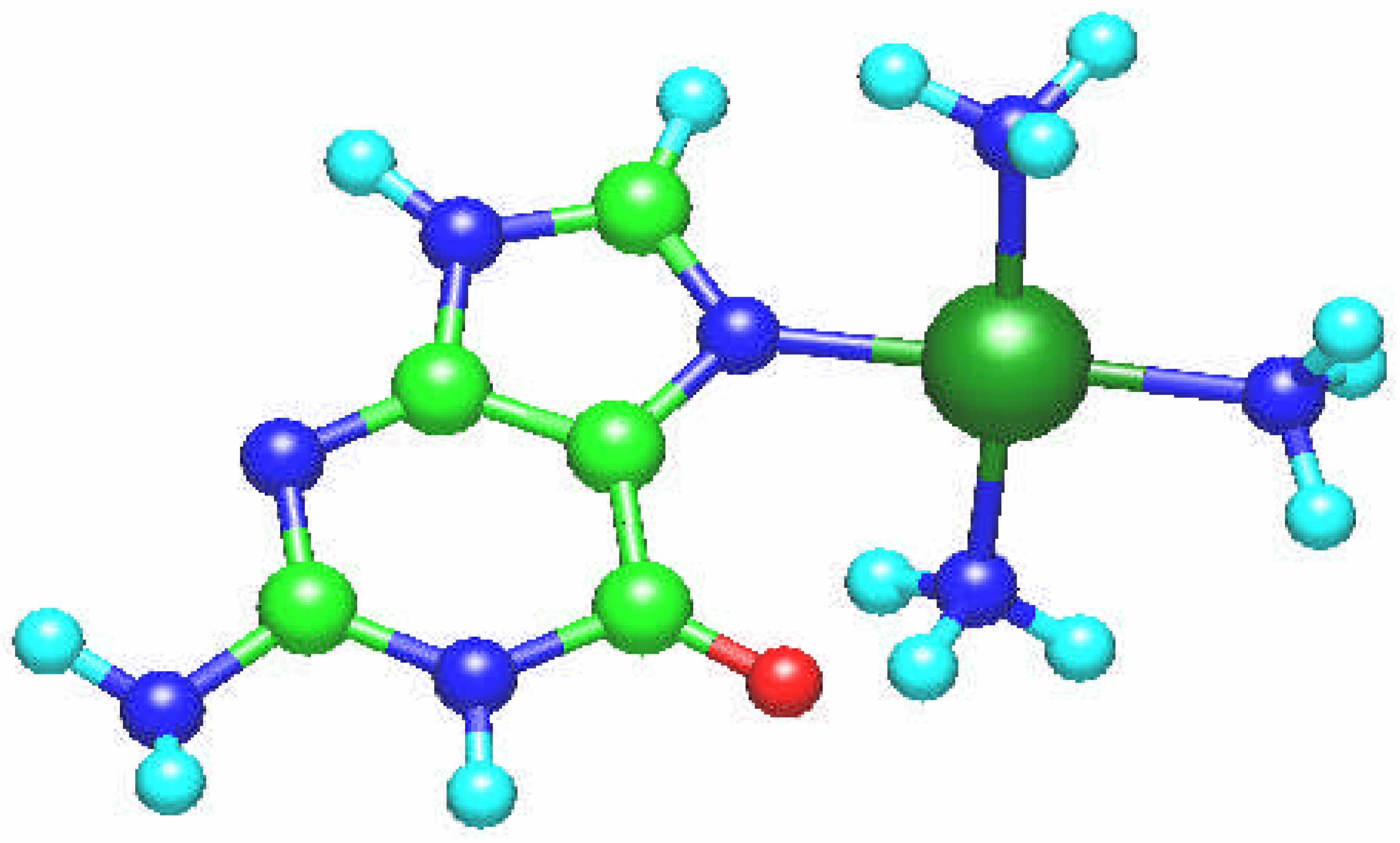
In the [Pt(NH
3)
2(guanine)]
2+ complex the two NH
3 ligands are coordinated in the plane of the guanine ring (
Fig. 1) and the cation is bonded to the N7 atom even when we start from the structure involving guanine ligand coordinated via N7 and O6 atoms. It means that the second bond breaks during the optimization process. This structure is different from other calculations [
19]. However, there may also occur some hydrogen bonding stabilization effect as the result of interaction of C – O … H (i.e. with the proton of the ammine group where O … H distance is of 1.05 Å). This result indicates that platinum via free coordination site can bind with other N atom forming intrastrand or interstrand crosslinks [
7] being one of possible modes of the specific interaction. Rather essential deformation of the guanine molecule was found with the –NH
2 group being out of the molecular plane. The ab initio Pt – N7 bond length is 2.002 Å and for the Pt – N amounts to 2.122 Å, whereas the distance Pt – O6 is equal to 2.663 Å and O-Pt-N angle is 109
o and 92
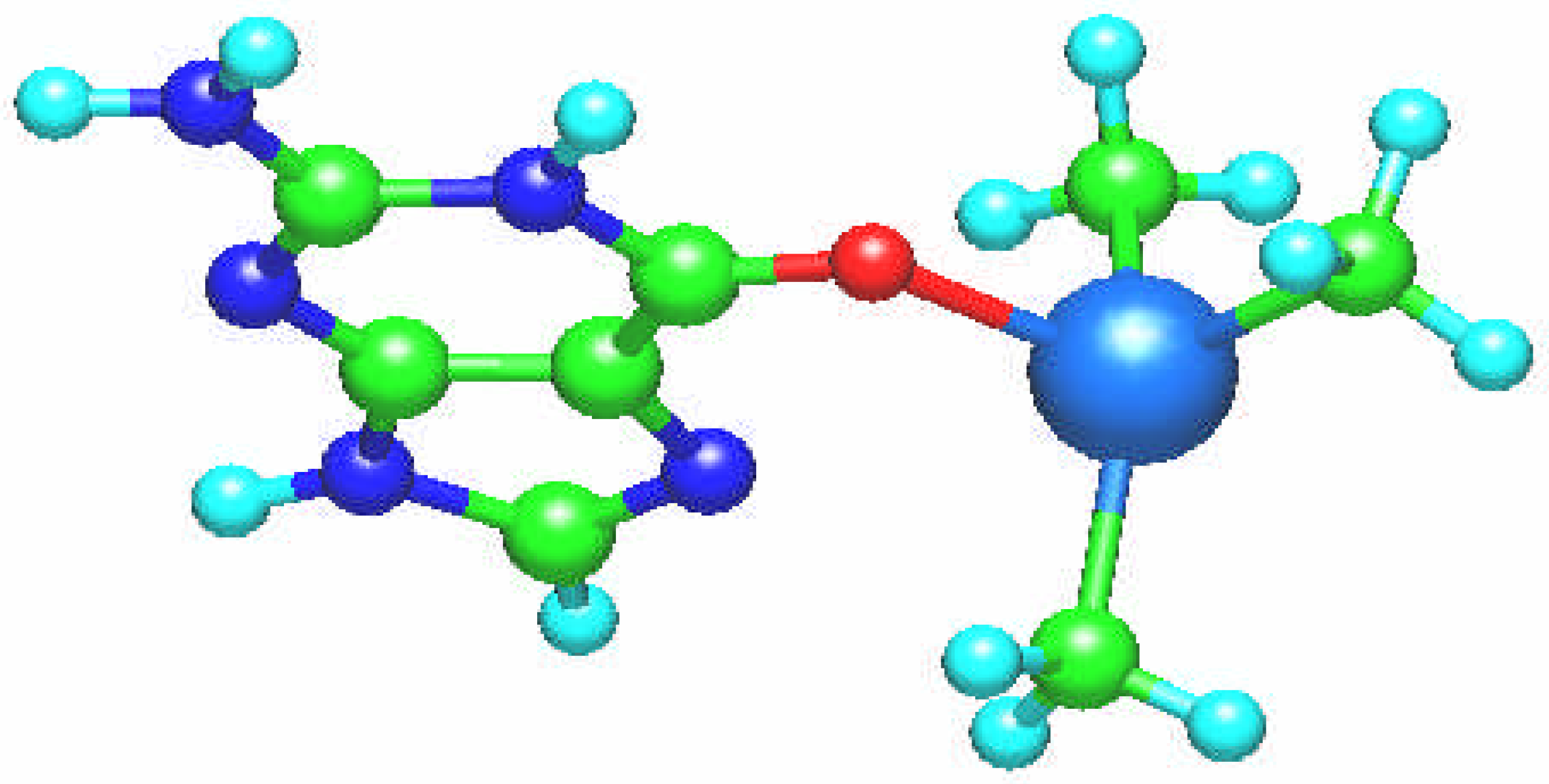
o, thus this complex has T shape. The platinum atom charge q(Pt), according to the Mulliken population, amounts to 1.304.
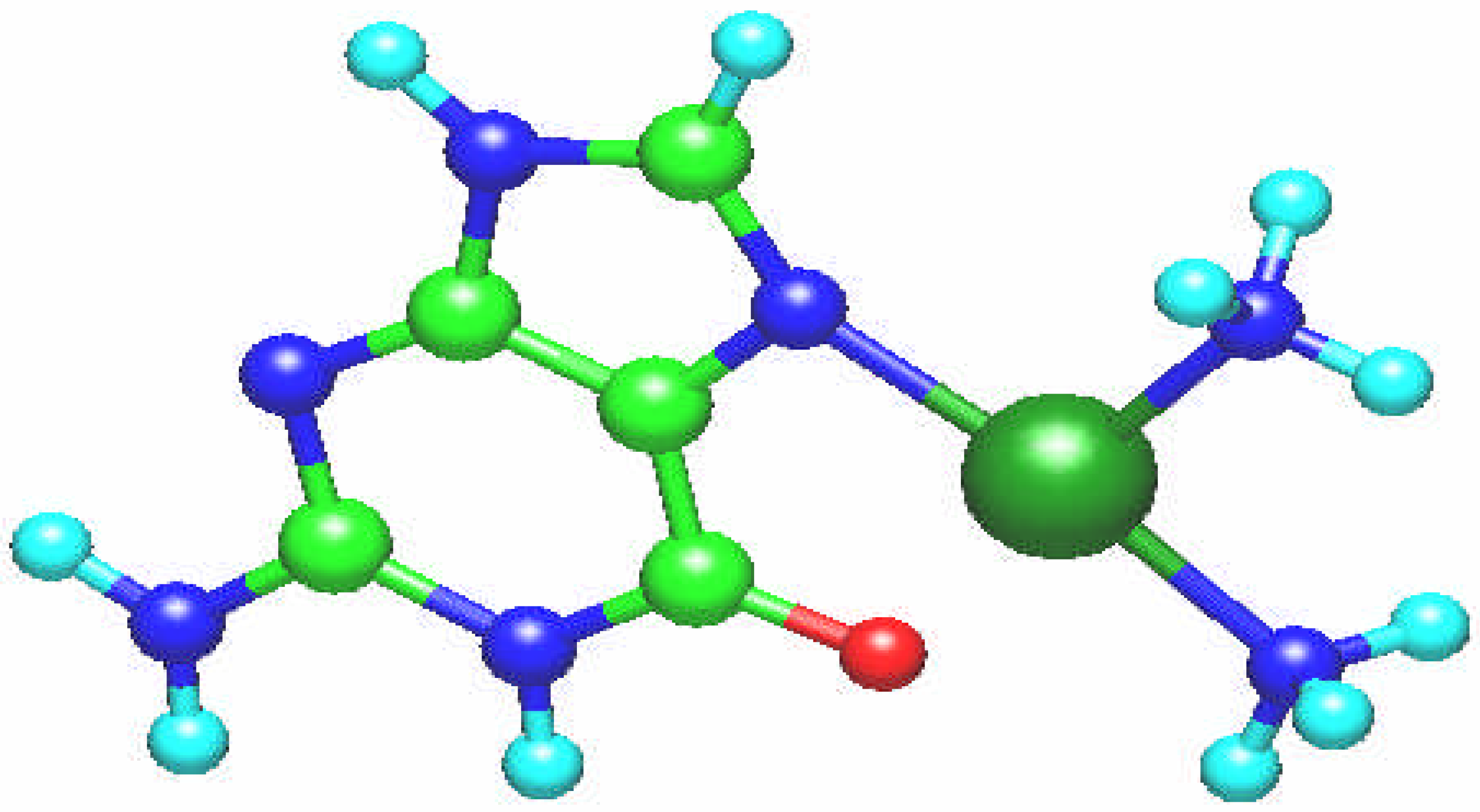
Fig. 1.
Molecular structure of the [Pt(NH3)2(guanine)]2+ complex.
Fig. 1.
Molecular structure of the [Pt(NH3)2(guanine)]2+ complex.
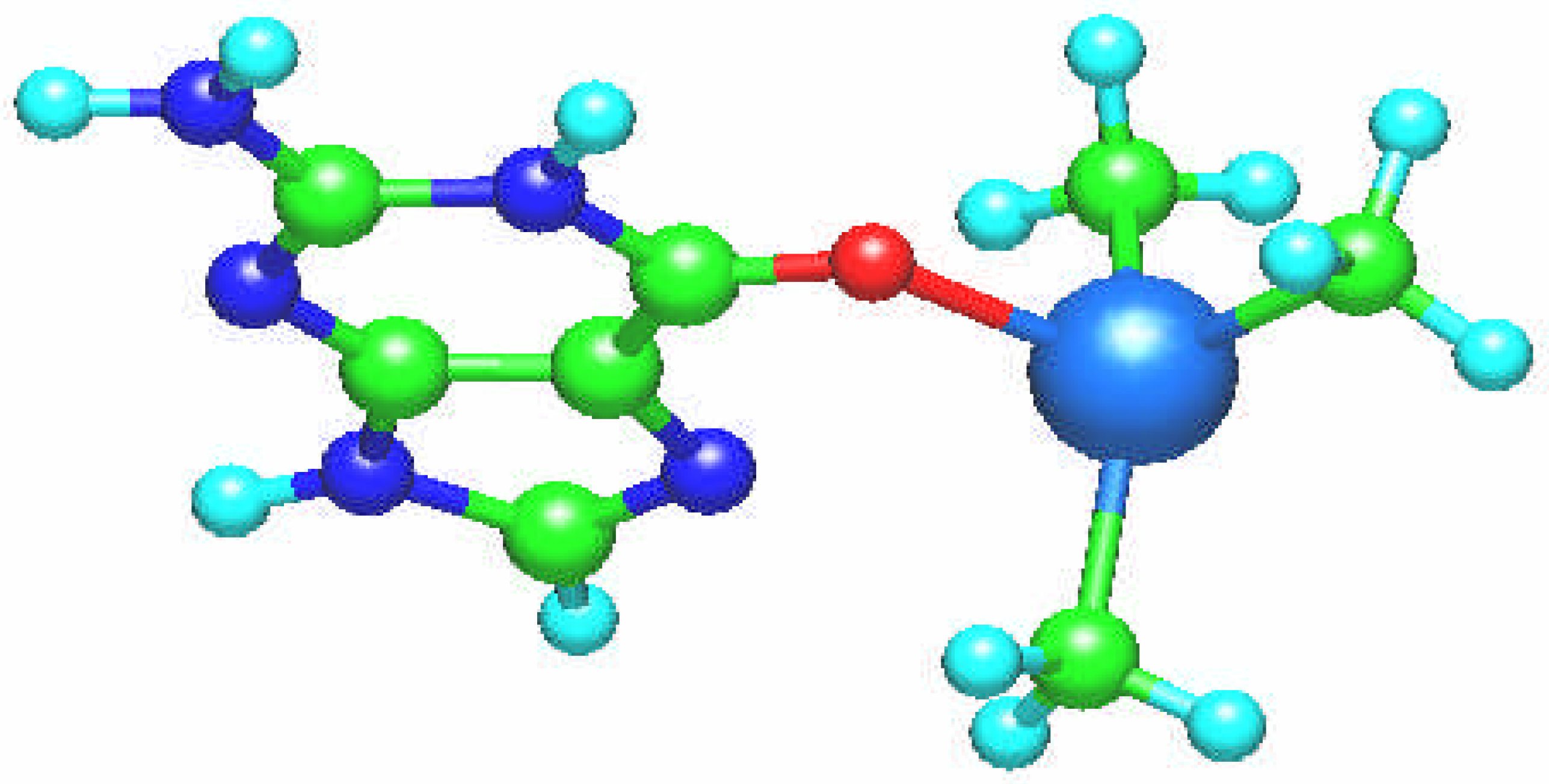
Fig. 2.
Molecular structure of the [Sn(CH3)2(guanine)]2+ complex.
Fig. 2.
Molecular structure of the [Sn(CH3)2(guanine)]2+ complex.
In the case of the dimethyltin [Sn(CH
3)
2(guanine)]
2+ complex guanine ligand is coordinated through N7 and O6 atoms as a chelate ligand (
Fig. 2). The tin complex has distorted tetrahedral coordination with C1-Sn-C2, N-Sn-C1, N-Sn-C2, O-Sn-C1, O-Sn-C2, and O-Sn - N angles are equal from 106
o to 112
o. The bond lengths are: Sn – N7: 2.201 Å, Sn – O6: 2.113 Å, Sn – C: 2.105 Å and the tin atomic charge is q(Sn) = 1.632.
However, interaction of the [Sn(CH
3)
2]
2+ cation with guanine can lead to the unexpected product, the complex in which one methyl group is transferrred to the O6 atom of the guanine molecule (
Fig. 3). This stable conformation was found when optimizing geometry of this moiety at the all-valence PM3 level with one CH
3 group connected to the O6 atom and Sn(CH
3)
2+ bonded covalently to the N7 atom (
Fig. 3). The ab initio binding energy for this conformation, which may be a transition complex, is greater by 10.23 eV in comparison with those for the structure depicted in
Fig. 2. In this case the the Sn – N7 bond length is 2.264 Å, and that of Sn-C(H
3) amounts to 2.150 Å. This result indicates that diorgano and triorganotin compounds can interact not only with nucleotide bases and phosphate groups of DNA, but also as an alkylating antitumor agents.
Table 2.
Binding energies [eV] for some model complexes of tin-DNA fragments
Table 2.
Binding energies [eV] for some model complexes of tin-DNA fragments
| COMPLEX | PM3 | AB INITIO |
|---|
| [Pt(NH3)2(guanine)]2+ | 11.13 | 5.86 |
| [Pt(NH3)3(guanine)]2+ | 5.92 | 5.63 |
| | | |
| [Sn(CH3)2(guanine)]2+ | 6.48 | 8.30 |
| [Sn(CH3)3(guanine)]+ | 2.28 | 3.62 |
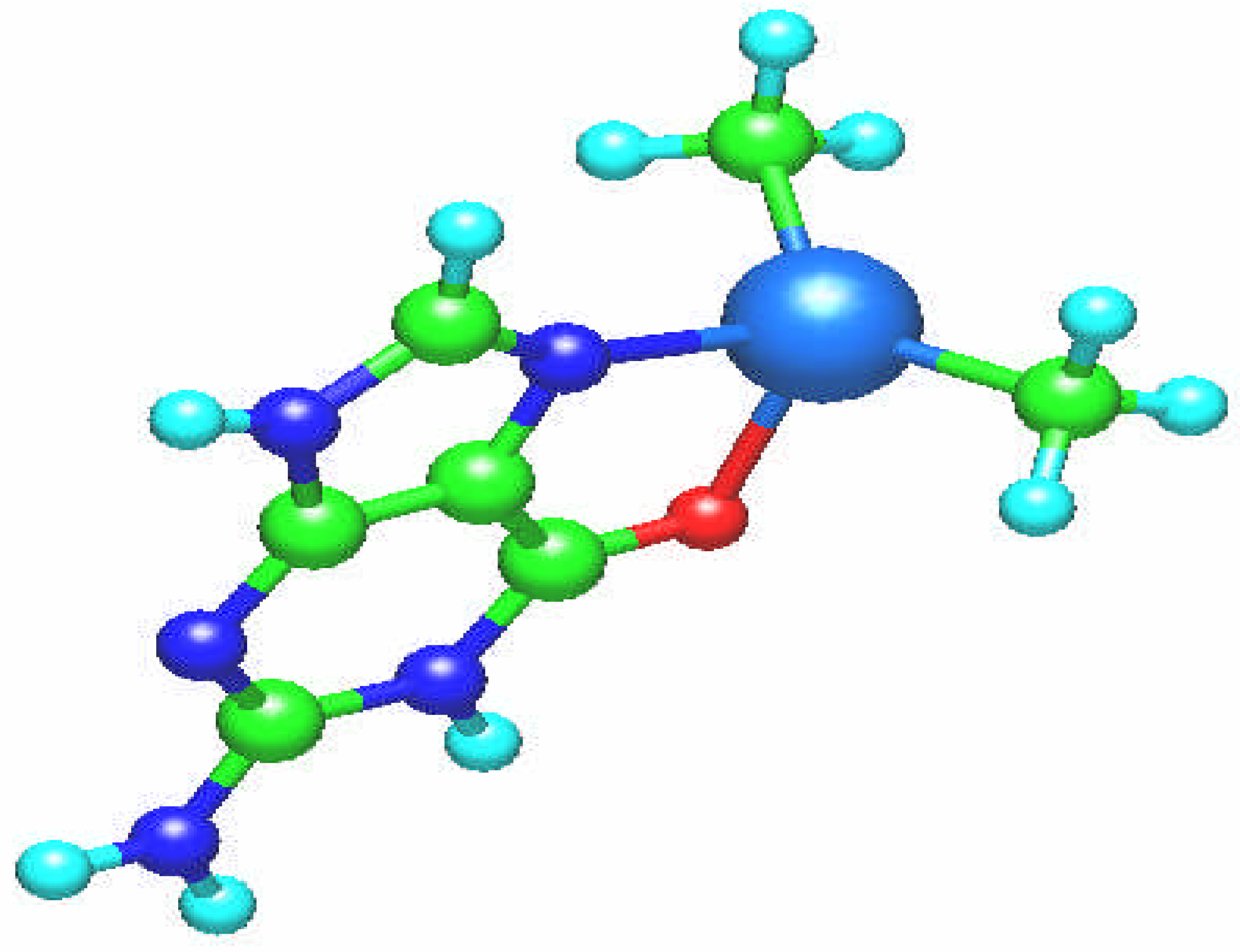
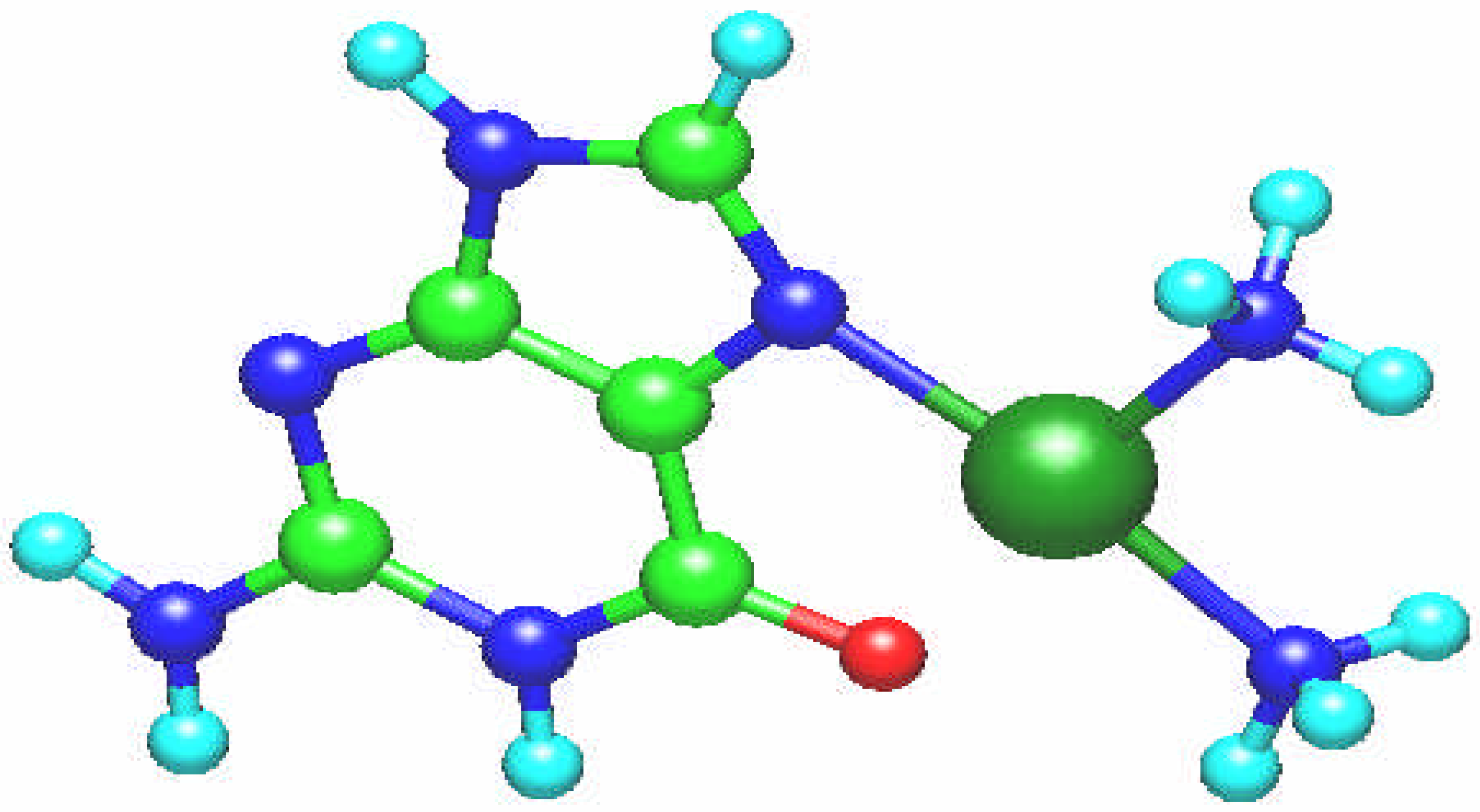
Fig. 3.
The unusual resulting molecular structure of the [Sn(CH3)2 (guanine)]2+ complex. One CH3 group is bonded to the oxygen O6 atom and [Sn(CH3)]2+ group to the nitrogen N7 atom in the ring plane.
Fig. 3.
The unusual resulting molecular structure of the [Sn(CH3)2 (guanine)]2+ complex. One CH3 group is bonded to the oxygen O6 atom and [Sn(CH3)]2+ group to the nitrogen N7 atom in the ring plane.
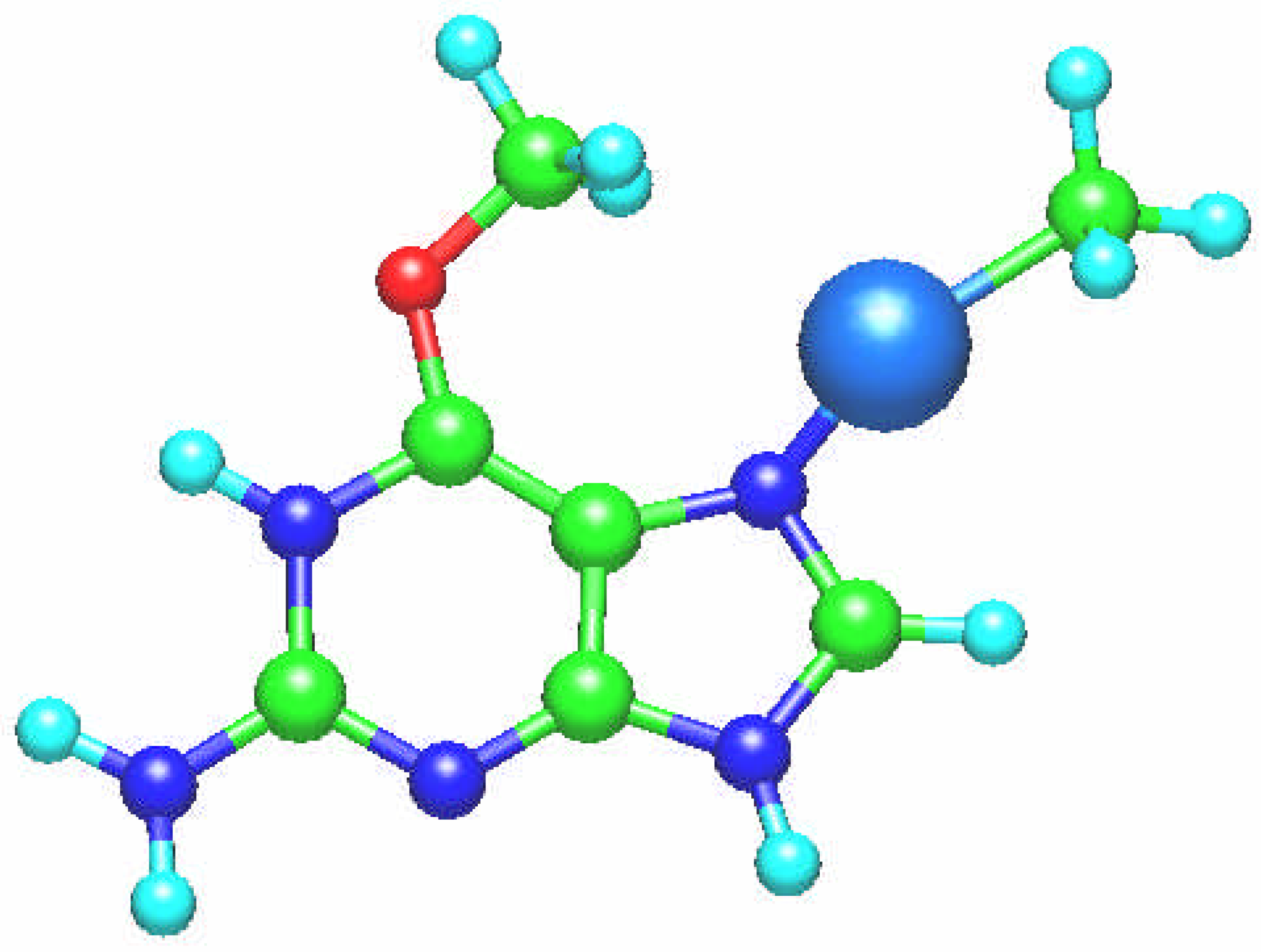
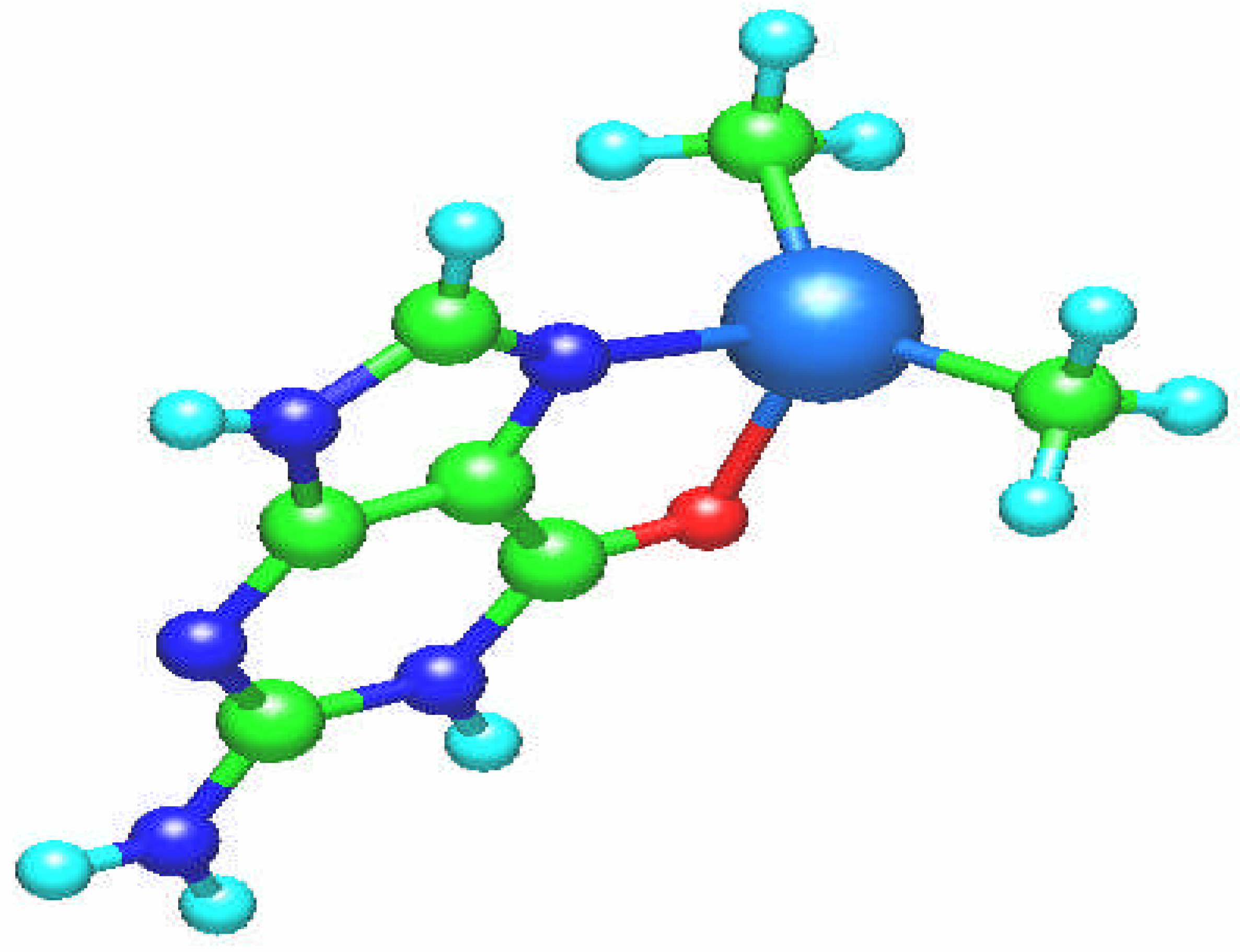
Fig. 4.
Molecular structure of the square planar [Pt(NH3)3(guanine)]2+ complex. The guanine ligand is perpendicular to the coordination plane.
Fig. 4.
Molecular structure of the square planar [Pt(NH3)3(guanine)]2+ complex. The guanine ligand is perpendicular to the coordination plane.
The [Pt(NH
3)
3(guanine)]
2+ complex is square planar with guanine ligand perpendicular to the coordination plane (
Fig. 4). The Pt – N bond length amounts to 2.004 Å and the platinum atom charge is 1.027.
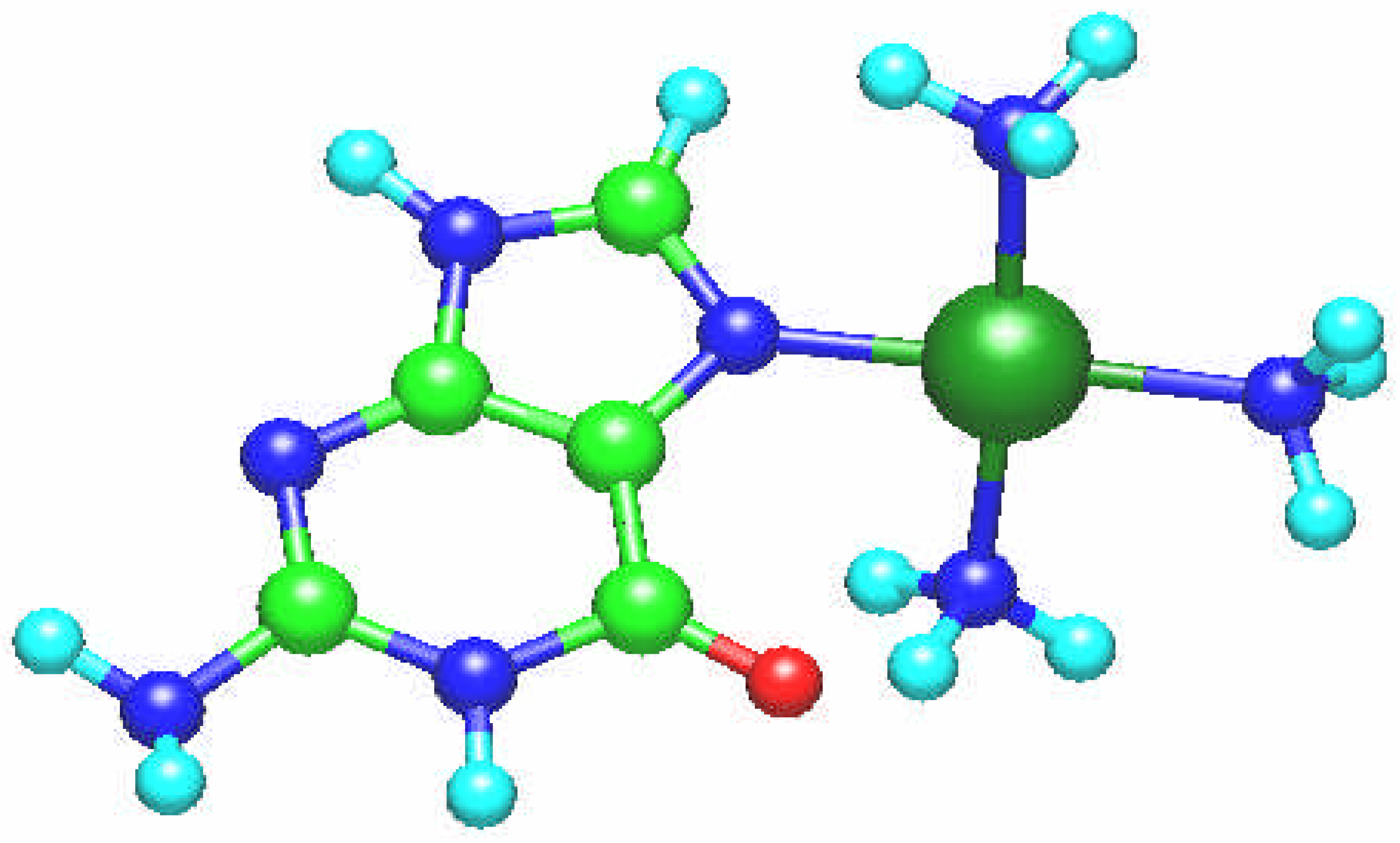
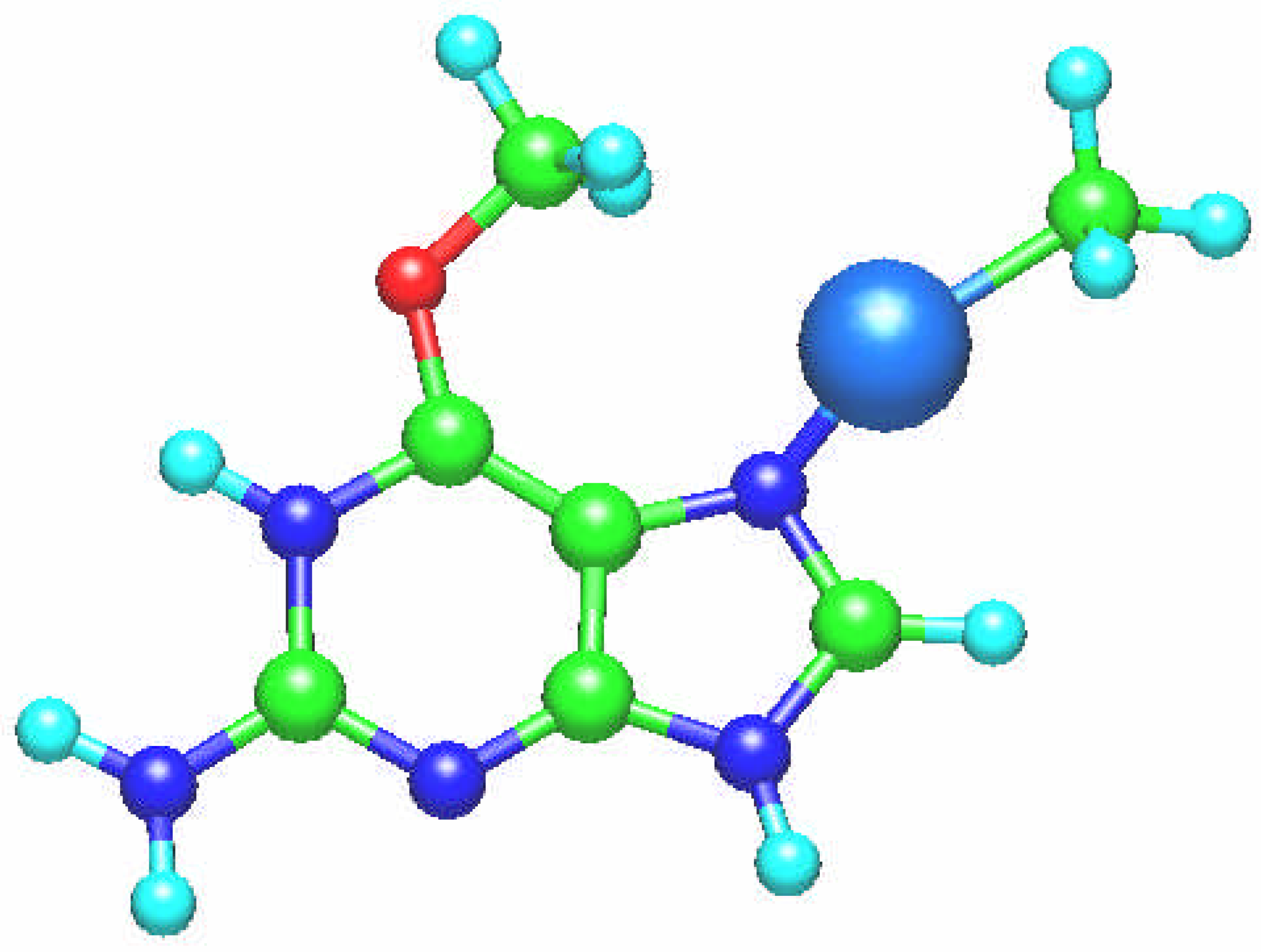
In the [Sn(CH
3)
3(guanine)]
+ complex (
Fig. 5) the guanine ligand is coordinated via O6 atom. In this case deviation from the tetrahedral coordination is not strong, the tetrahedral angles change in the range of 103
o – 112
o. The hydrogen bonding stabilizing effect is possible here as the interatomic distance N7 … H (of the –CH
3) group is 2.143 Å.
Fig. 5.
Molecular structure of the [Sn(NH3)3(guanine)]+ complex.
Fig. 5.
Molecular structure of the [Sn(NH3)3(guanine)]+ complex.
The results of calculations point out on a rather large binding energy for the organotin(IV) and platinum(II) complexes with guanine (
Table 1). The same was found for the interaction of the above cations with the phosphate anion H
2PO
4- both at the semiempirical and ab initio level. In the latter case the binding energy for [Pt(NH
3)
2(H2PO
4]
+ is 14.02 eV and in the case of [Sn(CH
3)
2(H
2 PO
4]
+ amounts to 16.57 eV.
The results of calculations given in this report seem to be reasonable in comparison with some of those attainable in the literature [
18]. In our case they were obtained at the correlation level influencing essentially the final results. Although the solvent effect has not been taken into account, the results still may be conclusive for further considerations of the mechanism of the tin derivatives acting as an antitumor agents. On the other hand, the role of water molecules was shown to play an important role [
24]. Anyway, it should be noted that the more detail explanation of the possible mechanism of the tin compounds acting as the antitumor drugs is a much more complicated problem.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}