Imidazo[1,2-b]pyrazole-7-Carboxamide Derivative Induces Differentiation-Coupled Apoptosis of Immature Myeloid Cells Such as Acute Myeloid Leukemia and Myeloid-Derived Suppressor Cells

 ,
,
Abstract
:
1. Introduction
2. Results
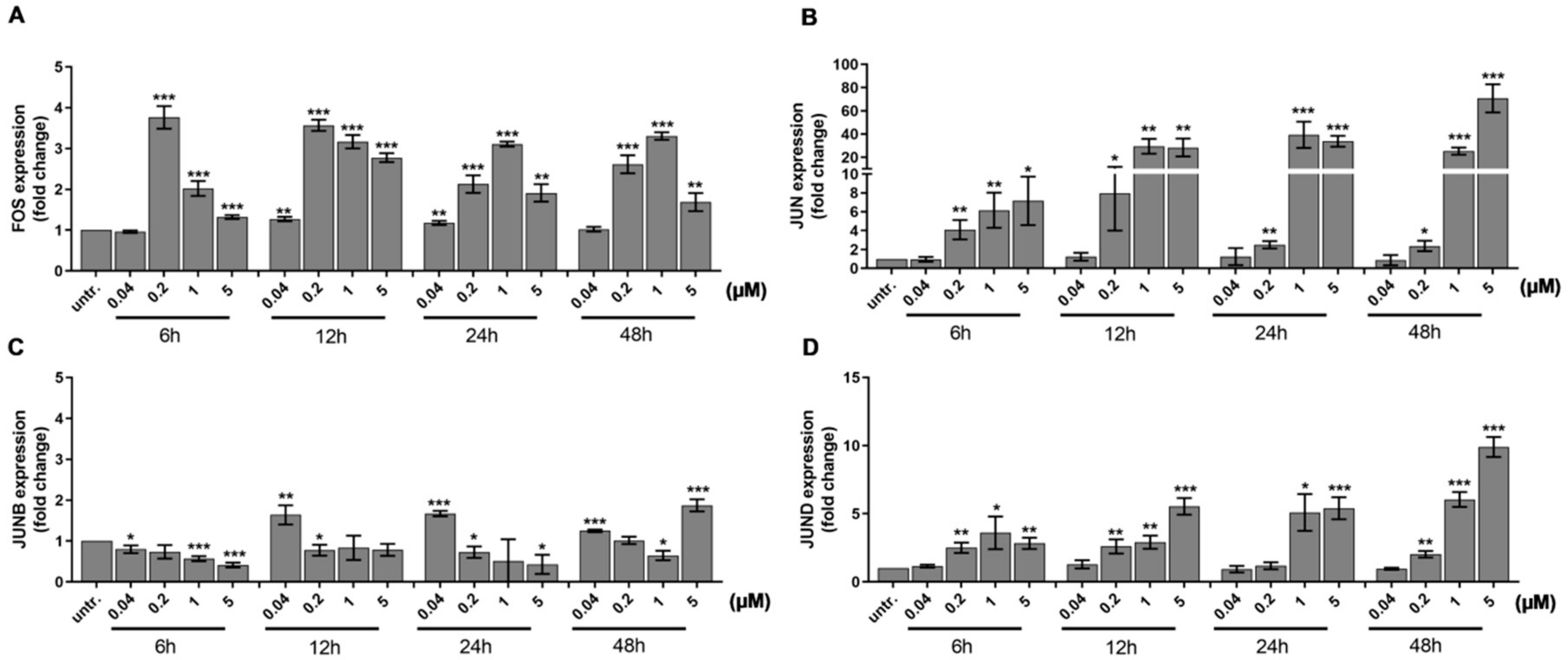
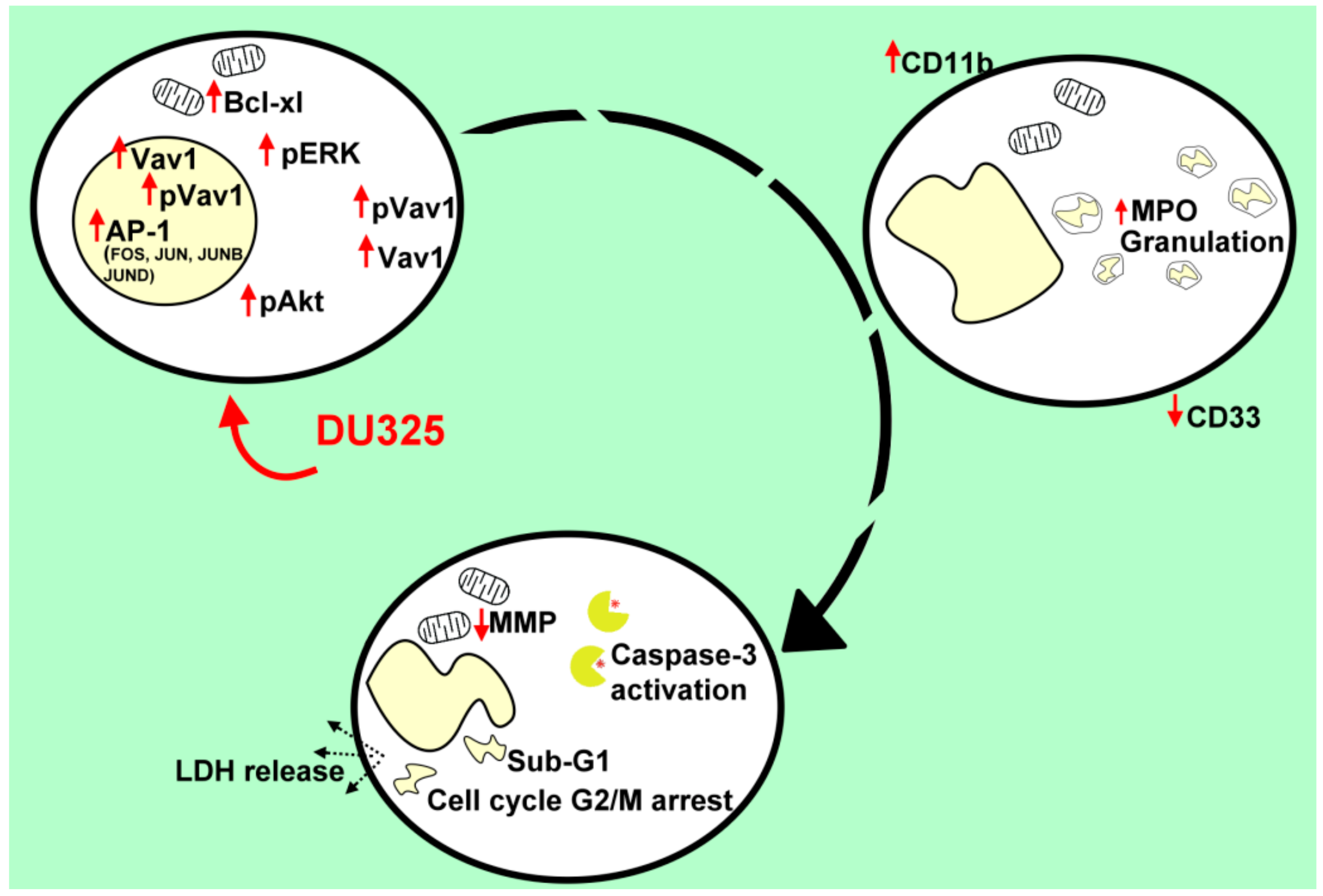
2.1. DU325 Drives Survival Pathways as an Early Response to Treatment in HL-60 Cells
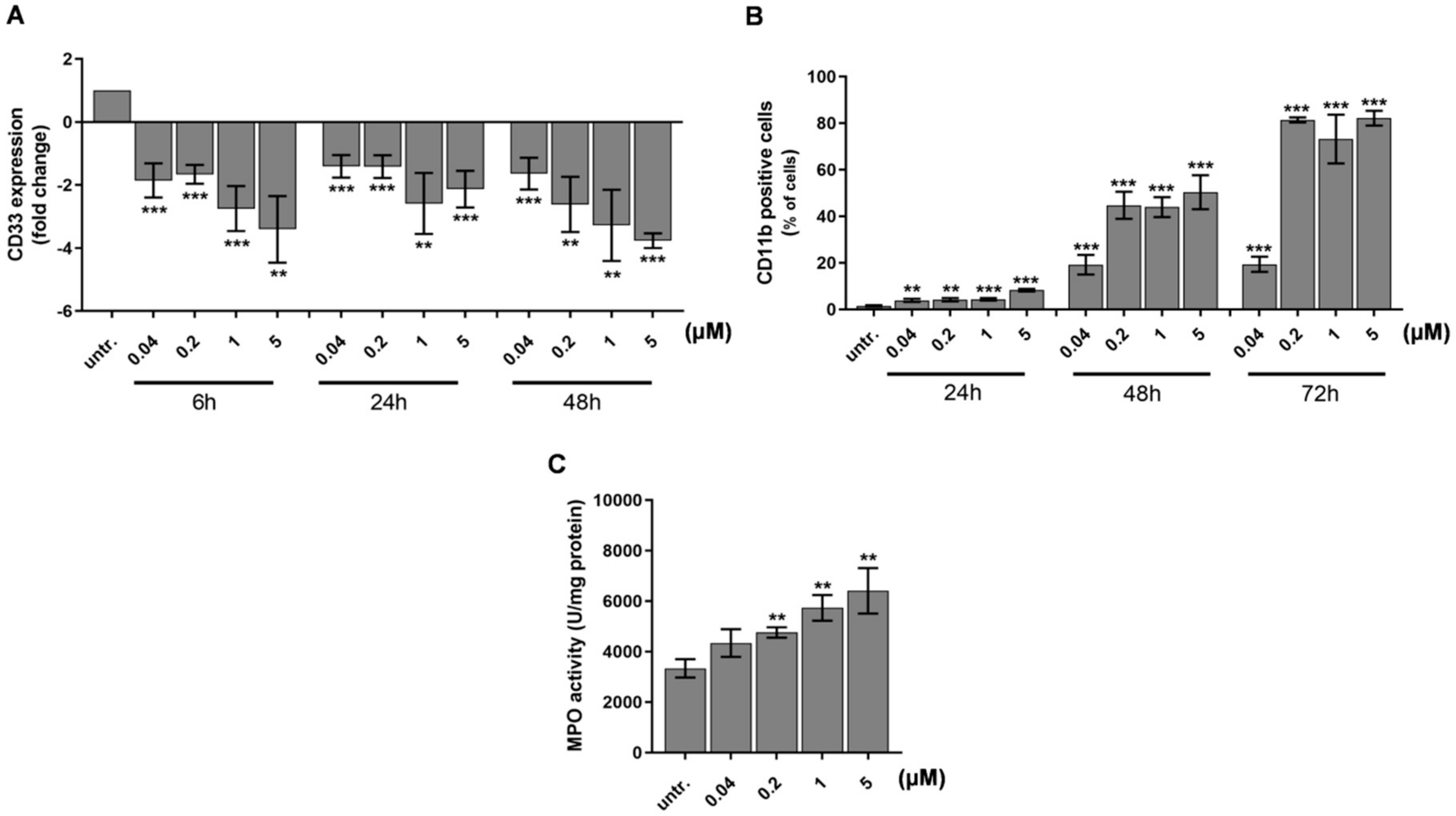
2.2. DU325 Induces Differentiation of HL-60 Cells
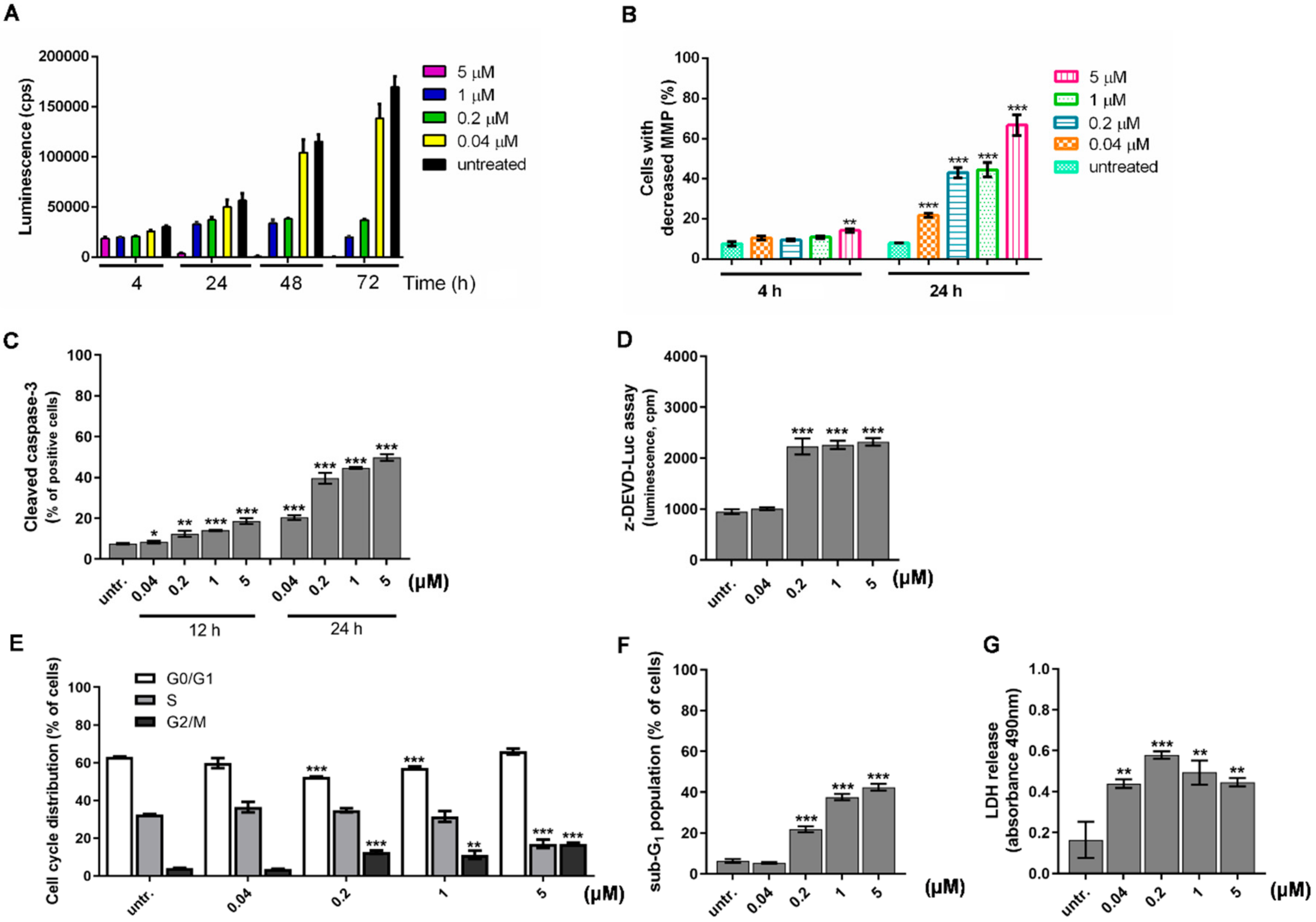
2.3. Differentiation of HL-60 Cells Is Followed by Apoptotic Cell Death
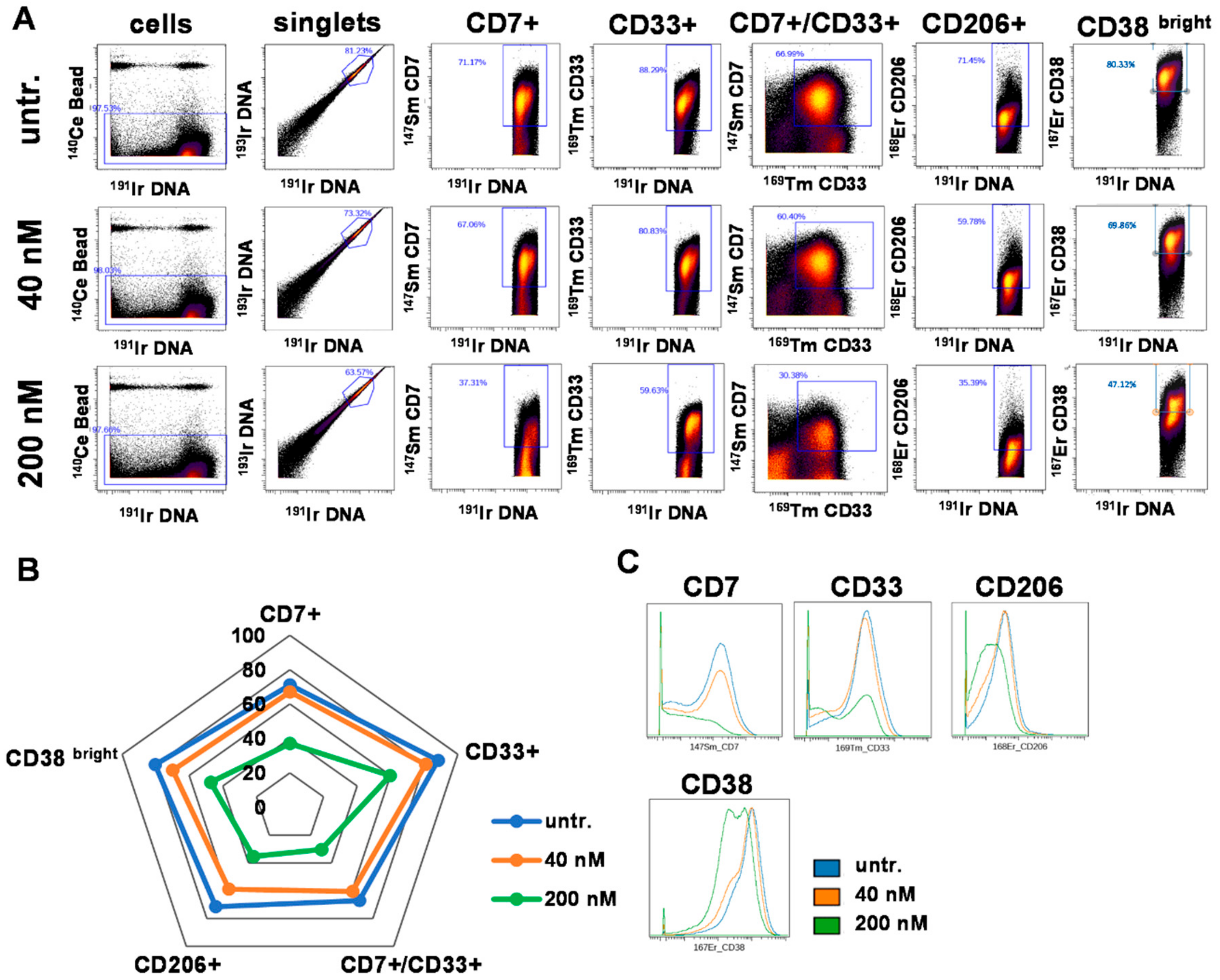
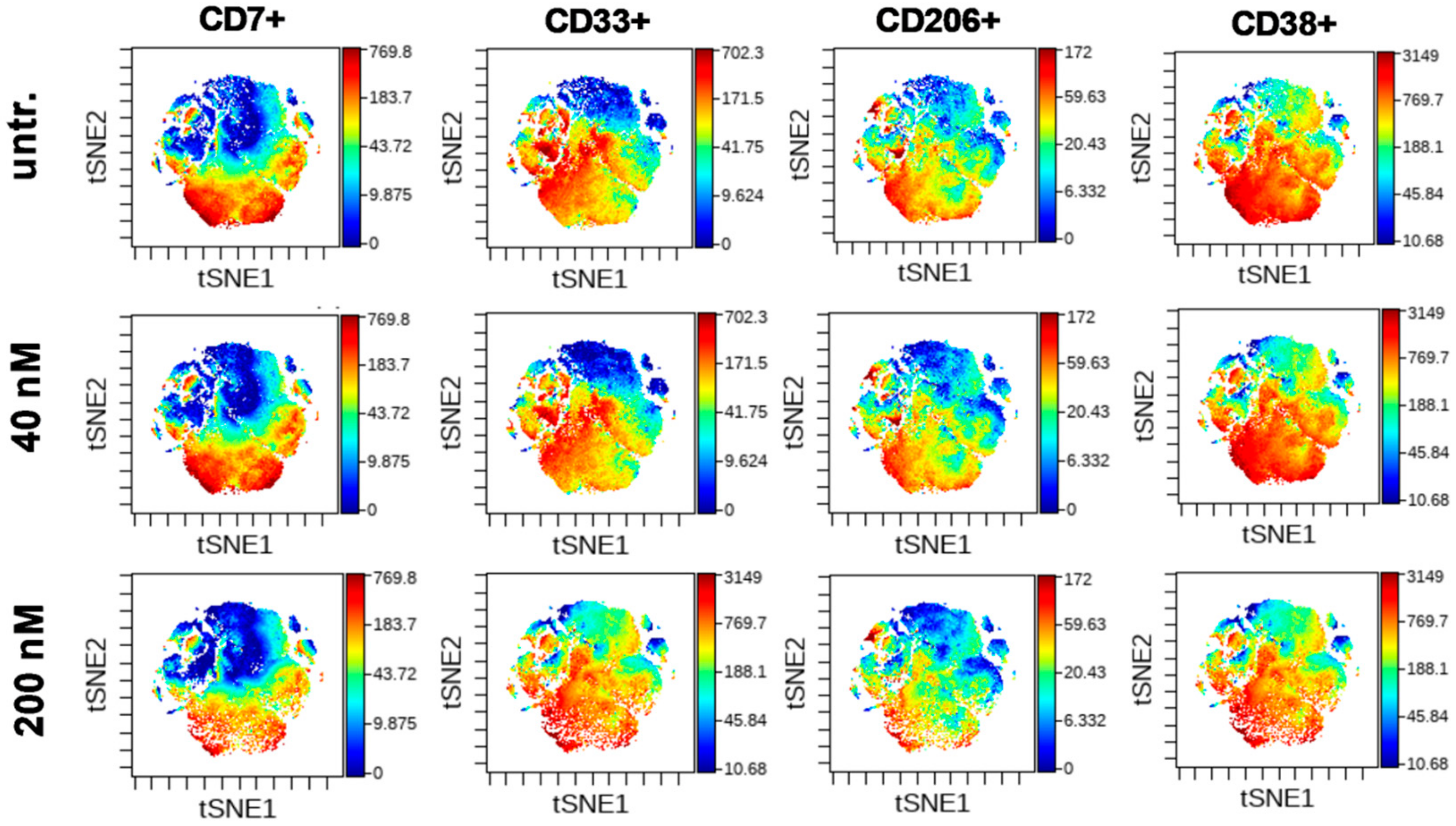
2.4. DU325 Induces Differentiation and Apoptosis of Human Primary AML Cells
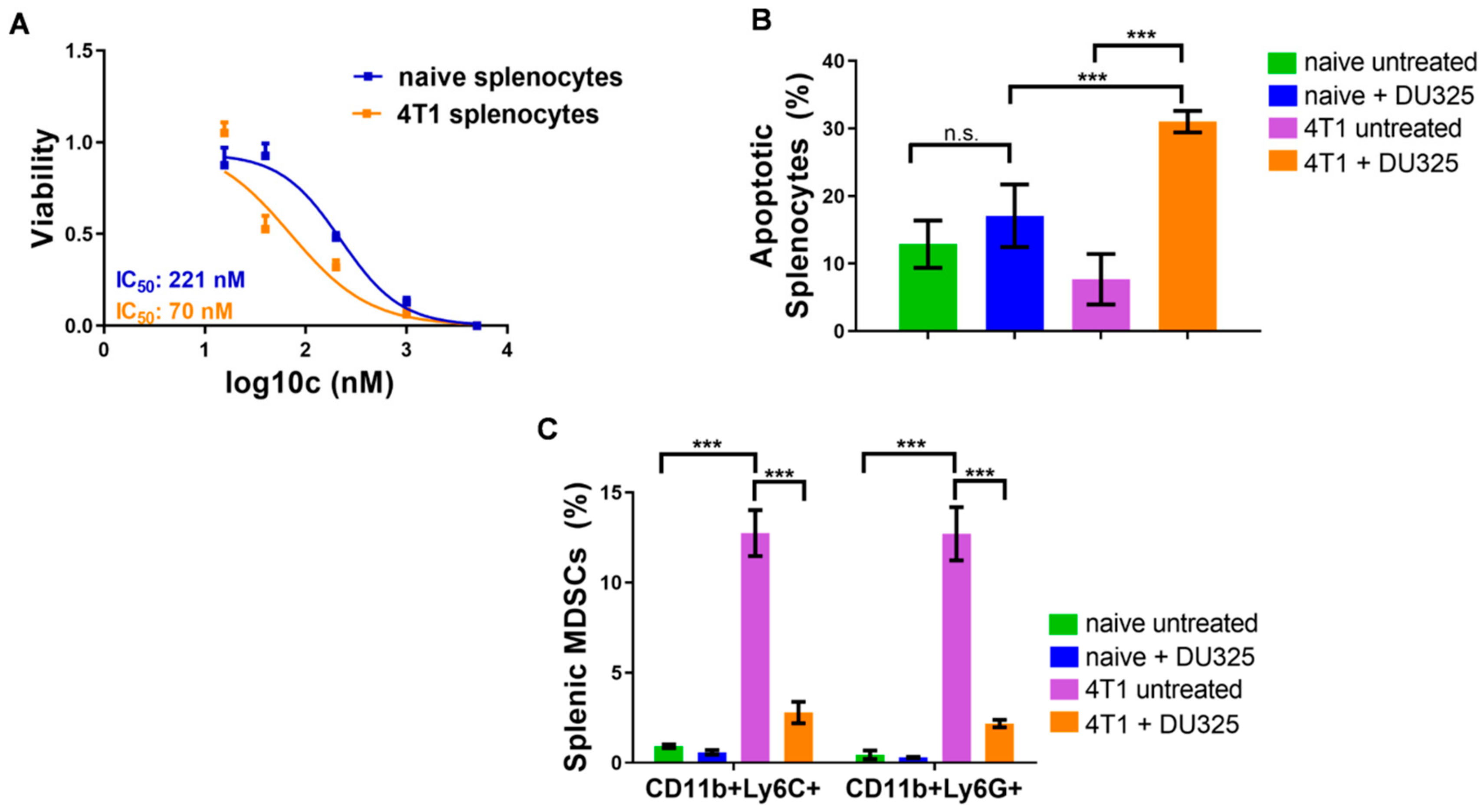
2.5. DU325 Treatment Ex Vivo Reduced Both Monocytic and Granulocytic Splenic MDSCs from the 4T1 Murine Breast Cancer Model
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Cell Culture and Isolation of Human AML Cells
4.3. Isolation of Murine Myeloid-Derived Suppressor Cells
4.4. Treatment by the Imidazo[1,2-b]pyrazole-7-carboxamide Derivative, DU325
4.5. Resazurin Viability Assay
4.6. RealTime-GloTM MT Cell Viability Assay
4.7. Z-DEVD-aLuc Caspase Assay
4.8. LDH Assay
4.9. Quantitative Real-Time PCR (qRT-PCR)
4.10. Purification of Nuclei and Immunochemical Analysis
4.11. Measurement of the MPO Activity
4.12. Fluorescence Flow Cytometry
4.12.1. Detection of the Mitochondrial Membrane Potential (MMP)
4.12.2. Detection of Phosphatidylserine Exposure
4.12.3. Cell Cycle and Sub-G1 Analysis
4.12.4. Immunofluorescence
4.13. Mass Cytometry
4.14. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germing, U.; Kobbe, G.; Haas, R.; Gattermann, N. Myelodysplastic syndromes: Diagnosis, prognosis, and treatment. Dtsch. Arztebl. Int. 2013, 110, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.M.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Garcia-Manero, G.; Pierce, S.; Nazha, A.; Bueso-Ramos, C.; Jabbour, E.; Ravandi, F.; Cortes, J.; Kantarjian, H. Interactions and relevance of blast percentage and treatment strategy among younger and older patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Am. J. Hematol. 2016, 91, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef]
- Moen, M.D.; McKeage, K.; Plosker, G.L.; Siddiqui, M.A. Imatinib: A review of its use in chronic myeloid leukaemia. Drugs 2007, 67, 299–320. [Google Scholar] [CrossRef]
- Gangat, N.; Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes: Contemporary review and how we treat. Am. J. Hematol. 2016, 91, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.; Nimer, S. Recent advances in the understanding and treatment of acute myeloid leukemia. F1000Res 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. ‘Acute myeloid leukemia: A comprehensive review and 2016 update’. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Chale, R.S.; Abad, A.C.; Schally, A.V. Acute promyelocytic leukemia (APL): A review of the literature. Oncotarget 2020, 11, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Birnie, G.D. The HL60 cell line: A model system for studying human myeloid cell differentiation. Br. J. Cancer Suppl. 1988, 9, 41–45. [Google Scholar] [PubMed]
- Mark Welch, D.B.; Jauch, A.; Langowski, J.; Olins, A.L.; Olins, D.E. Transcriptomes reflect the phenotypes of undifferentiated, granulocyte and macrophage forms of HL-60/S4 cells. Nucleus 2017, 8, 222–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Robertson, K.A.; Mueller, L. Retinoic acid-induced granulocytic differentiation of HL-60 myeloid leukemia cells is mediated directly through the retinoic acid receptor (RAR-alpha). Mol. Cell. Biol. 1990, 10, 2154–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.J.; Ruscetti, F.W.; Gallagher, R.E.; Gallo, R.C. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc. Natl. Acad. Sci. USA 1978, 75, 2458–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovera, G.; Santoli, D.; Damsky, C. Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc. Natl Acad. Sci. USA 1979, 76, 2779–2783. [Google Scholar] [CrossRef] [Green Version]
- Mangelsdorf, D.J.; Koeffler, H.P.; Donaldson, C.A.; Pike, J.W.; Haussler, M.R. 1,25-Dihydroxyvitamin D3-induced differentiation in a human promyelocytic leukemia cell line (HL-60): Receptor-mediated maturation to macrophage-like cells. J. Cell Biol. 1984, 98, 391–398. [Google Scholar] [CrossRef]
- Petrie, K.; Zelent, A.; Waxman, S. Differentiation therapy of acute myeloid leukemia: Past, present and future. Curr. Opin. Hematol. 2009, 16, 84–91. [Google Scholar] [CrossRef]
- De The, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Demjen, A.; Alfoldi, R.; Angyal, A.; Gyuris, M.; Hackler, L., Jr.; Szebeni, G.J.; Wolfling, J.; Puskas, L.G.; Kanizsai, I. Synthesis, cytotoxic characterization, and SAR study of imidazo[1,2-b]pyrazole-7-carboxamides. Arch. Pharm. (Weinh.) 2018, 351, e1800062. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Balog, J.A.; Demjen, A.; Alfoldi, R.; Vegi, V.L.; Feher, L.Z.; Man, I.; Kotogany, E.; Guban, B.; Batar, P.; et al. Imidazo[1,2-b]pyrazole-7-carboxamides Induce Apoptosis in Human Leukemia Cells at Nanomolar Concentrations. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting "Emergency" Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediat. Inflamm. 2017, 2017, 9294018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, H.; Watanabe, M. Myeloid-Derived Suppressor Cells and Therapeutic Strategies in Cancer. Mediat. Inflamm. 2015, 2015, 159269. [Google Scholar] [CrossRef] [Green Version]
- Shipp, C.; Speigl, L.; Janssen, N.; Martens, A.; Pawelec, G. A clinical and biological perspective of human myeloid-derived suppressor cells in cancer. Cell. Mol. Life Sci. 2016, 73, 4043–4061. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskas, L.G. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int. J. Mol. Sci. 2016, 17, 1958. [Google Scholar] [CrossRef]
- Balog, J.A.; Hackler, L., Jr.; Kovacs, A.K.; Neuperger, P.; Alfoldi, R.; Nagy, L.I.; Puskas, L.G.; Szebeni, G.J. Single Cell Mass Cytometry Revealed the Immunomodulatory Effect of Cisplatin Via Downregulation of Splenic CD44+, IL-17A+ MDSCs and Promotion of Circulating IFN-gamma+ Myeloid Cells in the 4T1 Metastatic Breast Cancer Model. Int. J. Mol. Sci. 2019, 21, 170. [Google Scholar] [CrossRef] [Green Version]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—a new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Masciulli, R.; Tritarelli, E.; Pustorino, R.; Mariani, G.; Martucci, R.; Barberi, T.; Camagna, A.; Valtieri, M.; Peschle, C. Transforming growth factor-beta potentiates vitamin D3-induced terminal monocytic differentiation of human leukemic cell lines. J. Immunol. 1993, 150, 2418–2430. [Google Scholar] [PubMed]
- Young, M.R.; Ihm, J.; Lozano, Y.; Wright, M.A.; Prechel, M.M. Treating tumor-bearing mice with vitamin D3 diminishes tumor-induced myelopoiesis and associated immunosuppression, and reduces tumor metastasis and recurrence. Cancer Immunol Immunother. 1995, 41, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Park, C.S.; Lee, Y.R.; Im, S.A.; Song, S.; Lee, C.K. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharm. Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef]
- Tasseff, R.; Jensen, H.A.; Congleton, J.; Dai, D.; Rogers, K.V.; Sagar, A.; Bunaciu, R.P.; Yen, A.; Varner, J.D. An Effective Model of the Retinoic Acid Induced HL-60 Differentiation Program. Sci. Rep. 2017, 7, 14327. [Google Scholar] [CrossRef] [Green Version]
- Bertagnolo, V.; Brugnoli, F.; Grassilli, S.; Nika, E.; Capitani, S. Vav1 in differentiation of tumoral promyelocytes. Cell Signal. 2012, 24, 612–620. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, L.; Ran, Q.; Wang, J.; Wang, C.; He, H.; Li, L.; Qi, H. Notopterol-induced apoptosis and differentiation in human acute myeloid leukemia HL-60 cells. Drug Des. Devel. Ther. 2019, 13, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E. Src family kinases and the MEK/ERK pathway in the regulation of myeloid differentiation and myeloid leukemogenesis. Adv. Enzym. Regul. 2008, 48, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Moon, D.O.; Asami, Y.; Kim, M.O.; Jang, J.H.; Kim, B.Y.; Ahn, J.S.; Kim, G.Y.; Yun, S.G. Xestospongin C induces monocytic differentiation of HL60 cells through activation of the ERK pathway. Food Chem. Toxicol. 2013, 55, 505–512. [Google Scholar] [CrossRef]
- Chatterjee, D.; Han, Z.; Mendoza, J.; Goodglick, L.; Hendrickson, E.A.; Pantazis, P.; Wyche, J.H. Monocytic differentiation of HL-60 promyelocytic leukemia cells correlates with the induction of Bcl-xL. Cell Growth Differ. 1997, 8, 1083–1089. [Google Scholar] [PubMed]
- Miranda, M.B.; Dyer, K.F.; Grandis, J.R.; Johnson, D.E. Differential activation of apoptosis regulatory pathways during monocytic vs granulocytic differentiation: A requirement for Bcl-X(L)and XIAP in the prolonged survival of monocytic cells. Leukemia 2003, 17, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busca, A.; Saxena, M.; Iqbal, S.; Angel, J.; Kumar, A. PI3K/Akt regulates survival during differentiation of human macrophages by maintaining NF-kappaB-dependent expression of antiapoptotic Bcl-xL. J. Leukoc. Biol. 2014, 96, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolo, V.; Marchisio, M.; Brugnoli, F.; Bavelloni, A.; Boccafogli, L.; Colamussi, M.L.; Capitani, S. Requirement of tyrosine-phosphorylated Vav for morphological differentiation of all-trans-retinoic acid-treated HL-60 cells. Cell Growth Differ. 2001, 12, 193–200. [Google Scholar]
- Bertagnolo, V.; Nika, E.; Brugnoli, F.; Bonora, M.; Grassilli, S.; Pinton, P.; Capitani, S. Vav1 is a crucial molecule in monocytic/macrophagic differentiation of myeloid leukemia-derived cells. Cell Tissue Res. 2011, 345, 163–175. [Google Scholar] [CrossRef]
- Houlard, M.; Arudchandran, R.; Regnier-Ricard, F.; Germani, A.; Gisselbrecht, S.; Blank, U.; Rivera, J.; Varin-Blank, N. Vav1 is a component of transcriptionally active complexes. J. Exp. Med. 2002, 195, 1115–1127. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Ku, G.; Weiss, A. A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J. Biol. Chem. 2000, 275, 2185–2190. [Google Scholar] [CrossRef] [Green Version]
- Brugnoli, F.; Lambertini, E.; Varin-Blank, N.; Piva, R.; Marchisio, M.; Grassilli, S.; Miscia, S.; Capitani, S.; Bertagnolo, V. Vav1 and PU.1 are recruited to the CD11b promoter in APL-derived promyelocytes: Role of Vav1 in modulating PU.1-containing complexes during ATRA-induced differentiation. Exp. Cell Res. 2010, 316, 38–47. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C.; Tugores, A.; Flores, I.; Naranjo, J.R. Differences in expression of transcription factor AP-1 in human promyelocytic HL-60 cells during differentiation towards macrophages versus granulocytes. Biochem. J. 1993, 294, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [Green Version]
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to AP-1. J. Leukoc. Biol. 1998, 63, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminuma, O.; Deckert, M.; Elly, C.; Liu, Y.C.; Altman, A. Vav-Rac1-mediated activation of the c-Jun N-terminal kinase/c-Jun/AP-1 pathway plays a major role in stimulation of the distal NFAT site in the interleukin-2 gene promoter. Mol. Cell. Biol. 2001, 21, 3126–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Rudd, C.E. CD28 and Grb-2, relative to Gads or Grap, preferentially co-operate with Vav1 in the activation of NFAT/AP-1 transcription. Biochem Biophys Res. Commun. 2008, 369, 616–621. [Google Scholar] [CrossRef] [Green Version]
- Doyle, B.T.; O’Neill, A.J.; Newsholme, P.; Fitzpatrick, J.M.; Watson, R.W. The loss of IAP expression during HL-60 cell differentiation is caspase-independent. J. Leukoc. Biol. 2002, 71, 247–254. [Google Scholar]
- Jensen, H.A.; Yourish, H.B.; Bunaciu, R.P.; Varner, J.D.; Yen, A. Induced myelomonocytic differentiation in leukemia cells is accompanied by noncanonical transcription factor expression. FEBS Open Bio 2015, 5, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Doyle, B.T.; O’Neill, A.J.; Fitzpatrick, J.M.; Watson, R.W. Differentiation-induced HL-60 cell apoptosis: A mechanism independent of mitochondrial disruption? Apoptosis 2004, 9, 345–352. [Google Scholar] [CrossRef]
- Duellman, S.J.; Zhou, W.; Meisenheimer, P.; Vidugiris, G.; Cali, J.J.; Gautam, P.; Wennerberg, K.; Vidugiriene, J. Bioluminescent, Nonlytic, Real-Time Cell Viability Assay and Use in Inhibitor Screening. Assay Drug Dev. Technol. 2015, 13, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018. [Google Scholar] [CrossRef]
- Amir el, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Moschovi, M.; Critselis, E.; Cen, O.; Adamaki, M.; Lambrou, G.I.; Chrousos, G.P.; Vlahopoulos, S. Drugs acting on homeostasis: Challenging cancer cell adaptation. Expert Rev. Anticancer Ther. 2015, 15, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Winer, E.S.; Stone, R.M. Novel therapy in Acute myeloid leukemia (AML): Moving toward targeted approaches. Ther. Adv. Hematol. 2019, 10, 2040620719860645. [Google Scholar] [CrossRef]
- Bertagnolo, V.; Grassilli, S.; Petretto, A.; Lambertini, E.; Astati, L.; Bruschi, M.; Brugnoli, F.; Nika, E.; Candiano, G.; Piva, R.; et al. Nuclear proteome analysis reveals a role of Vav1 in modulating RNA processing during maturation of tumoral promyelocytes. J. Proteom. 2011, 75, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Kuranda, K.; Berthon, C.; Lepretre, F.; Polakowska, R.; Jouy, N.; Quesnel, B. Expression of CD34 in hematopoietic cancer cell lines reflects tightly regulated stem/progenitor-like state. J. Cell. Biochem. 2011, 112, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.M.; Allen, L.A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell. Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.D.; Voyich, J.M.; Somerville, G.A.; Braughton, K.R.; Malech, H.L.; Musser, J.M.; DeLeo, F.R. An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J. Leukoc. Biol. 2003, 73, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.W.; Rotstein, O.D.; Parodo, J.; Bitar, R.; Hackam, D.; Marshall, J.C. Granulocytic differentiation of HL-60 cells results in spontaneous apoptosis mediated by increased caspase expression. FEBS Lett. 1997, 412, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell. Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Atilla, E.; Atilla, P.A.; Mo, F.; Tashiro, H.; Srinivasan, M.; Lulla, P.; Rouce, R.H.; Cabral, J.M.S.; Ramos, C.A.; et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol. Ther. 2019, 27, 272–280. [Google Scholar] [CrossRef]
- Walter, R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 339–348. [Google Scholar] [CrossRef]
- Naik, J.; Themeli, M.; de Jong-Korlaar, R.; Ruiter, R.W.J.; Poddighe, P.J.; Yuan, H.; de Bruijn, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica 2019, 104, 100–103. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gils, N.; Verhagen, H.; Smit, L. Reprogramming acute myeloid leukemia into sensitivity for retinoic-acid-driven differentiation. Exp. Hematol. 2017, 52, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, H.; van Gils, N.; Martianez, T.; van Rhenen, A.; Rutten, A.; Denkers, F.; de Leeuw, D.C.; Smit, M.A.; Tsui, M.L.; de Vos Klootwijk, L.L.E.; et al. IGFBP7 Induces Differentiation and Loss of Survival of Human Acute Myeloid Leukemia Stem Cells without Affecting Normal Hematopoiesis. Cell. Rep. 2018, 25, 3021–3035. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell. Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Mathias, B.; Delmas, A.L.; Ozrazgat-Baslanti, T.; Vanzant, E.L.; Szpila, B.E.; Mohr, A.M.; Moore, F.A.; Brakenridge, S.C.; Brumback, B.A.; Moldawer, L.L.; et al. Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann. Surg. 2017, 265, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Janols, H.; Bergenfelz, C.; Allaoui, R.; Larsson, A.M.; Ryden, L.; Bjornsson, S.; Janciauskiene, S.; Wullt, M.; Bredberg, A.; Leandersson, K. A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J. Leukoc. Biol. 2014, 96, 685–693. [Google Scholar] [CrossRef] [Green Version]
- McPeak, M.B.; Youssef, D.; Williams, D.A.; Pritchett, C.; Yao, Z.Q.; McCall, C.E.; El Gazzar, M. Myeloid Cell-Specific Knockout of NFI-A Improves Sepsis Survival. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [Green Version]
- Szebeni, G.J.; Kriston-Pal, E.; Blazso, P.; Katona, R.L.; Novak, J.; Szabo, E.; Czibula, A.; Fajka-Boja, R.; Hegyi, B.; Uher, F.; et al. Identification of galectin-1 as a critical factor in function of mouse mesenchymal stromal cell-mediated tumor promotion. PLoS ONE 2012, 7, e41372. [Google Scholar] [CrossRef] [Green Version]
- Fajka-Boja, R.; Szebeni, G.J.; Hunyadi-Gulyas, E.; Puskas, L.G.; Katona, R.L. Polyploid Adipose Stem Cells Shift the Balance of IGF1/IGFBP2 to Promote the Growth of Breast Cancer. Front. Oncol. 2020, 10, 157. [Google Scholar] [CrossRef] [Green Version]
- Szebeni, G.J.; Balazs, A.; Madarasz, I.; Pocz, G.; Ayaydin, F.; Kanizsai, I.; Fajka-Boja, R.; Alfoldi, R.; Hackler, L., Jr.; Puskas, L.G. Achiral Mannich-Base Curcumin Analogs Induce Unfolded Protein Response and Mitochondrial Membrane Depolarization in PANC-1 Cells. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.K.; Hegyes, P.; Szebeni, G.J.; Nagy, L.I.; Puskas, L.G.; Toth, G.K. Synthesis of N-peptide-6-amino-D-luciferin Conjugates. Front. Chem. 2018, 6, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szebeni, G.J.; Tancos, Z.; Feher, L.Z.; Alfoldi, R.; Kobolak, J.; Dinnyes, A.; Puskas, L.G. Real architecture For 3D Tissue (RAFT) culture system improves viability and maintains insulin and glucagon production of mouse pancreatic islet cells. Cytotechnology 2017, 69, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertagnolo, V.; Grassilli, S.; D’Aguanno, S.; Brugnoli, F.; Bavelloni, A.; Faenza, I.; Nika, E.; Urbani, A.; Cocco, L.; Capitani, S. Mass spectrometry-based identification of Y745 of Vav1 as a tyrosine residue crucial in maturation of acute promyelocytic leukemia-derived cells. J. Proteome Res. 2010, 9, 752–760. [Google Scholar] [CrossRef]
- Posa, A.; Szabo, R.; Csonka, A.; Veszelka, M.; Berko, A.M.; Barath, Z.; Menesi, R.; Pavo, I.; Gyongyosi, M.; Laszlo, F.; et al. Endogenous Estrogen-Mediated Heme Oxygenase Regulation in Experimental Menopause. Oxid. Med. Cell. Longev. 2015, 2015, 429713. [Google Scholar] [CrossRef]
- Alfoldi, R.; Balog, J.A.; Farago, N.; Halmai, M.; Kotogany, E.; Neuperger, P.; Nagy, L.I.; Feher, L.Z.; Szebeni, G.J.; Puskas, L.G. Single Cell Mass Cytometry of Non-Small Cell Lung Cancer Cells Reveals Complexity of In vivo And Three-Dimensional Models over the Petri-dish. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Product | Gene Symbol | Accession Number | Forward Primer | Reverse Primer |
|---|---|---|---|---|
| Myeloid Cell Surface Antigen CD33 | CD33 | NM_001082618.1 NM_001177608.1 | ctgacctgctctgtgtcctg | atgagcaccgaggagtgagt |
| Jun Proto-Oncogene | JUN | NM_002228.3 | ccaaaggatagtgcgatgttt | ctgtccctctccactgcaac |
| Fos Proto-Oncogene | FOS | NM_005252.3 | actaccactcacccgcagac | ccaggtccgtgcagaagt |
| JunB Proto-Oncogene | JUNB | NM_002229.2 | atacacagctacgggatacgg | gctcggtttcaggagtttgt |
| JunD Proto-Oncogene | JUND | NM_005354.5 | cagcgaggagcaggagtt | gagctggttctgcttgtgtaaat |
| Actin Beta | ACTB | BC002409.2 | attggcaatgagcggttc | cgtggatgccacaggact |
| Target | Host/Clone | Fluorochrome | Supplier, Cat. Number | Dilution in the Assay |
|---|---|---|---|---|
| pERK1/2 (Thr202/Tyr204) | Mouse mAb, 4B11B69 | Alexa Fluor® 488 | Biolegend, 675508 | 1:25 |
| Bcl-xl | Rabbit mAb, 54H6 | Alexa Fluor® 488 | Cell Sign. Techn. 2767 | 1:75 |
| pAkt (Ser473) | Rabbit mAb, D9E | Alexa Fluor® 488 | Cell Sign. Techn. 4071 | 1:50 |
| Caspase-3 (Asp175) | Rabbit polyclonal | none | Cell Sign. Techn. 9661 | 1:500 |
| Anti-rabbit (H+L) | Goat polyclonal | Alexa Fluor® 488 | Thermo Fisher Sci. A-11008 | 1:600 |
| CD11b | Mouse mAb, LT11 | FITC | ImmunoTools, 21389113 | 1:20 |
| CD33 | Mouse mAb, WM53 | PE-Cy5.5 | Thermo Fisher Sci. A15454 | 1:00 |
| CD45 | Mouse mAb, 30-F11 | FITC | Biolegend, 103107 | 1:200 |
| CD11b | Rat mAb, M1/70 | PE | Biolegend, 101208 | 1:80 |
| Ly6C | Rat mAb, HK1.4 | APC | Biolegend, 128016 | 1:80 |
| Ly6G | Rat mAb, 1A8 | APC | Biolegend, 127614 | 1:330 |
| Target | Clone | Metal Tag |
|---|---|---|
| CD19 | HIB19 | 142Nd |
| CD11b | ICRF44 | 144Nd |
| CD7 | CD7-6B7 | 147Sm |
| CD66 | CD66a-B1.1 | 149Sm |
| CD36 | 5-271 | 152Sm |
| CD163 | GHI/61 | 154Sm |
| CD45 | HI30 | 156Gd |
| CD11c | Bu15 | 159Tb |
| CD14 | M5E2 | 160Gd |
| CD16 | 3G8 | 165Ho |
| CD38 | HIT2 | 167Er |
| CD206 | 15-2 | 168Er |
| CD33 | WM53 | 169Tm |
| CD3 | UCHT1 | 170Er |
| HLA-DR | L243 | 174Yb |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotogány, E.; Balog, J.Á.; Nagy, L.I.; Alföldi, R.; Bertagnolo, V.; Brugnoli, F.; Demjén, A.; Kovács, A.K.; Batár, P.; Mezei, G.; et al. Imidazo[1,2-b]pyrazole-7-Carboxamide Derivative Induces Differentiation-Coupled Apoptosis of Immature Myeloid Cells Such as Acute Myeloid Leukemia and Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2020, 21, 5135. https://doi.org/10.3390/ijms21145135
Kotogány E, Balog JÁ, Nagy LI, Alföldi R, Bertagnolo V, Brugnoli F, Demjén A, Kovács AK, Batár P, Mezei G, et al. Imidazo[1,2-b]pyrazole-7-Carboxamide Derivative Induces Differentiation-Coupled Apoptosis of Immature Myeloid Cells Such as Acute Myeloid Leukemia and Myeloid-Derived Suppressor Cells. International Journal of Molecular Sciences. 2020; 21(14):5135. https://doi.org/10.3390/ijms21145135
Chicago/Turabian StyleKotogány, Edit, József Á. Balog, Lajos I. Nagy, Róbert Alföldi, Valeria Bertagnolo, Federica Brugnoli, András Demjén, Anita K. Kovács, Péter Batár, Gabriella Mezei, and et al. 2020. "Imidazo[1,2-b]pyrazole-7-Carboxamide Derivative Induces Differentiation-Coupled Apoptosis of Immature Myeloid Cells Such as Acute Myeloid Leukemia and Myeloid-Derived Suppressor Cells" International Journal of Molecular Sciences 21, no. 14: 5135. https://doi.org/10.3390/ijms21145135