3. Discussion
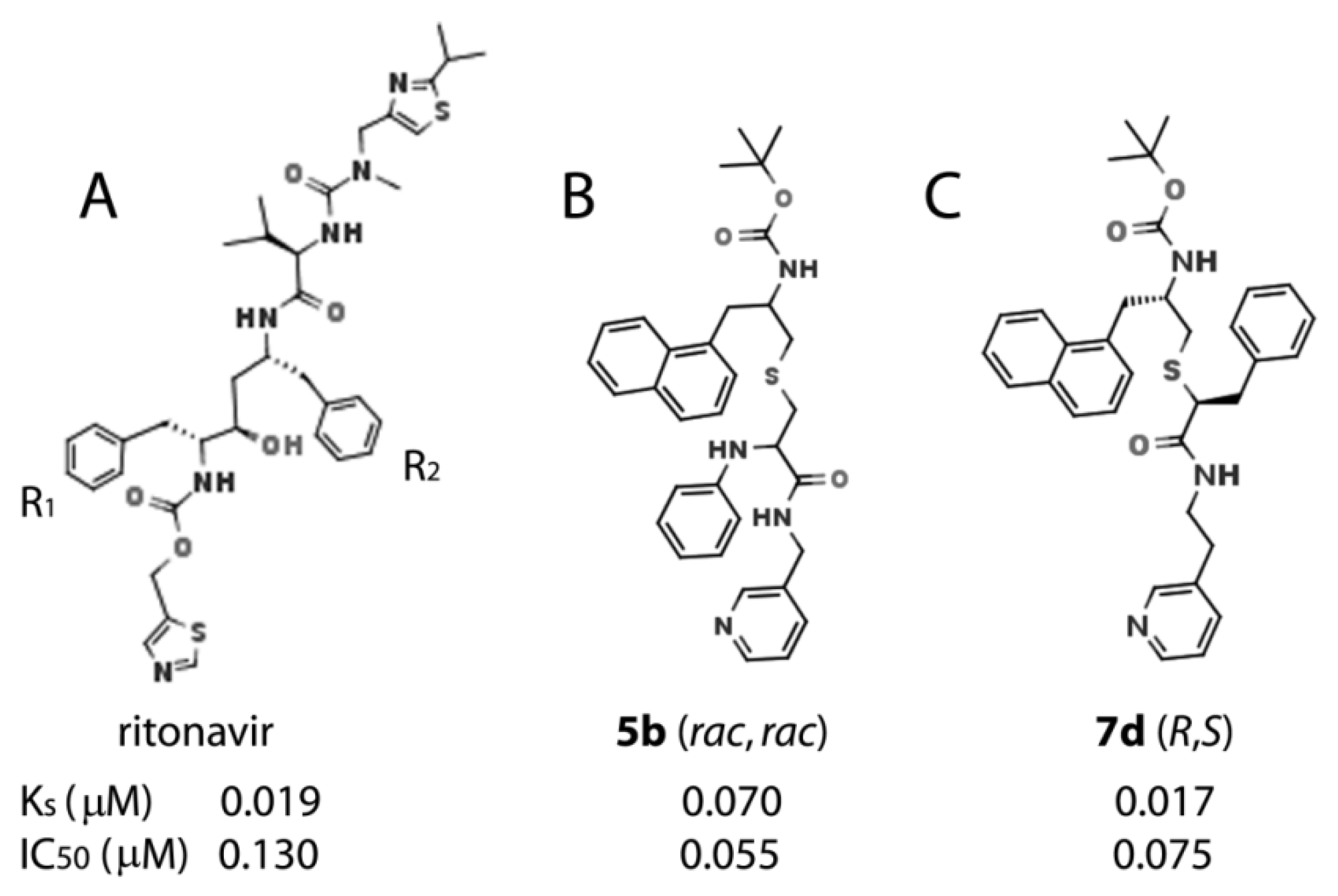
This work was a continuation of our efforts to identify structural attributes required for potent inhibition of human drug-metabolizing CYP3A4, the major and most clinically relevant P450 isoform. This knowledge is needed for a better understanding of the CYP3A4 inhibitory/ligand binding mechanism and building a pharmacophore model for a potent CYP3A4 inhibitor. The identification of an ensemble of electronic and steric features leading to potent CYP3A4 inhibition could help develop safer drugs and more effective pharmacoenhancers. Ritonavir, originally designed as an anti-HIV drug [
36], remains the most potent CYP3A4 inhibitor in clinical use [
37] and serves as a ‘gold standard’ in our studies. Based on the SAR analysis of the closest analogues of ritonavir, we constructed the first structure-based pharmacophore model for a potent CYP3A4-specific inhibitor [
19] and tested some of the determinants, including the backbone length/composition, R
1 and R
2 substituents, and pyridine–R
1 and R
1–R
2 spacers [
10,
20,
21,
22]. This study builds on our prior work and was performed to elucidate the optimal length of the pyridine–R
1 linker.
The linker extension from pyridyl–ethyl to pyridyl–propyl (six- to seven-atom pyridine–R
1 separation) was found to be more optimal for the R
1/R
2–phenyl containing compounds,
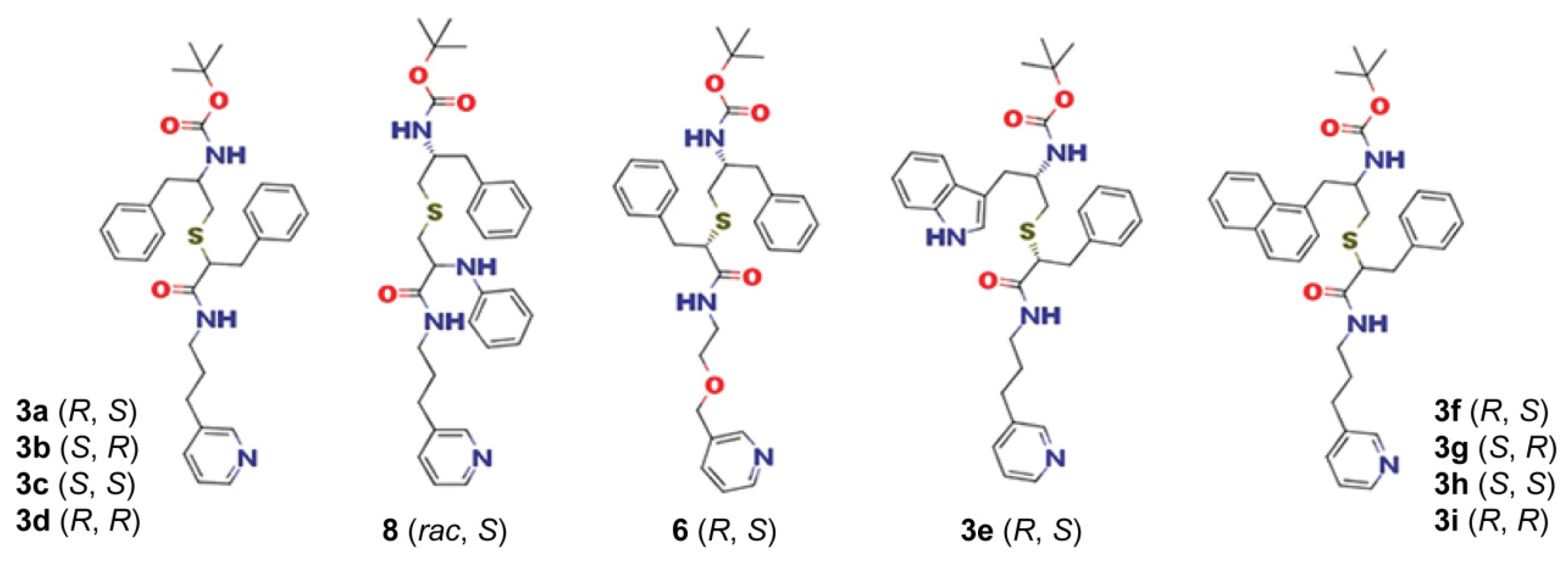
3a–
d, as it increased both the binding and inhibitory strength by several-fold (
Table 1). For the R
1–phenyl/R
2-indole/naphthalene analogues,
3e–
i, the impact was not uniform and depended on the side-group stereochemistry. In accordance with the previous findings [
10,
22], we confirmed that the
S/
R configuration was the least favourable, because it forces analogues to bind in reverse or sub-optimal orientations. In contrast, regardless of the scaffold,
R/
S was the most preferable conformation and, once again, was selected by CYP3A4 during co-crystallization with
8 (
rac,
S). The series V front-runner, however, was
3h (
S,
S). This analogue has outstandingly low K
s (0.007 μM) and inhibits CYP3A4 nearly as potently as the prior lead compounds (IC
50 of 0.090 μM vs. 0.055–0.070 μM for
5b and
7d) and 1.5-fold stronger than ritonavir (K
s and IC
50 of 0.019 and 0.130 μM, respectively).
The currently prevalent notion is that ritonavir acts as a mechanism-based inhibitor by producing reactive metabolite(s) inactivating CYP3A4 via covalent attachment [
38,
39,
40,
41,
42]. In our experimental system, we could not detect time-dependent CYP3A4 inactivation upon preincubation with NADPH and, instead, observed a slight decrease in IC
50 for ritonavir, likely due to its partial metabolism [
22]. Whether the membrane bilayer, cytochrome b
5 and/or other microsomal proteins are needed to mediate the reactive species production remains to be established. However, this and our prior studies demonstrate that the key factors contributing to the inhibition of CYP3A4 are the tight heme binding and blockage of the active site.
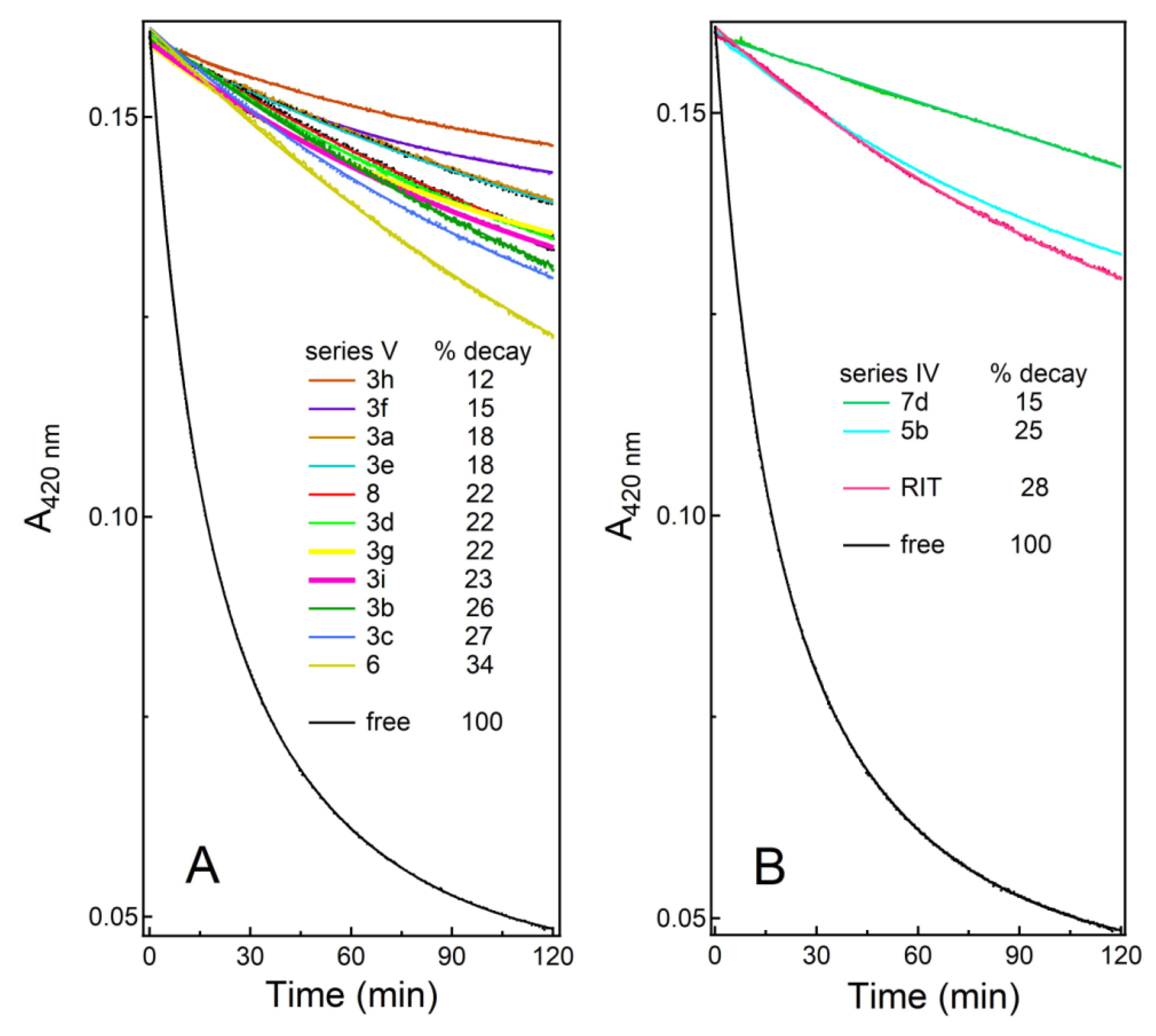
One problem with series V compounds was that, aside from the best and the worst inhibitors, the ranking was difficult due to low spread in IC
50 (
Table 1). By definition, IC
50 is the half inhibitory concentration, which under our experimental conditions (0.1 μM CYP3A4; 1:1 enzyme–inhibitor complex) is limited to 0.05 μM. This explains the IC
50 clustering and predicts a similar scenario for the next generation of optimized compounds. Contrarily, K
s does not depend on protein concentration and, as demonstrated for
3h, its significant improvement can still be achieved. The newly introduced assay, H
2O
2-dependent heme bleaching, helped to better evaluate the heme accessibility in the inhibitor-bound CYP3A4 and demonstrated that, compared to the best series IV and V analogues, the protective effect of ritonavir was considerably lower. This further underlines the importance of strong heme ligation and the fuller occupancy of the P2 site.
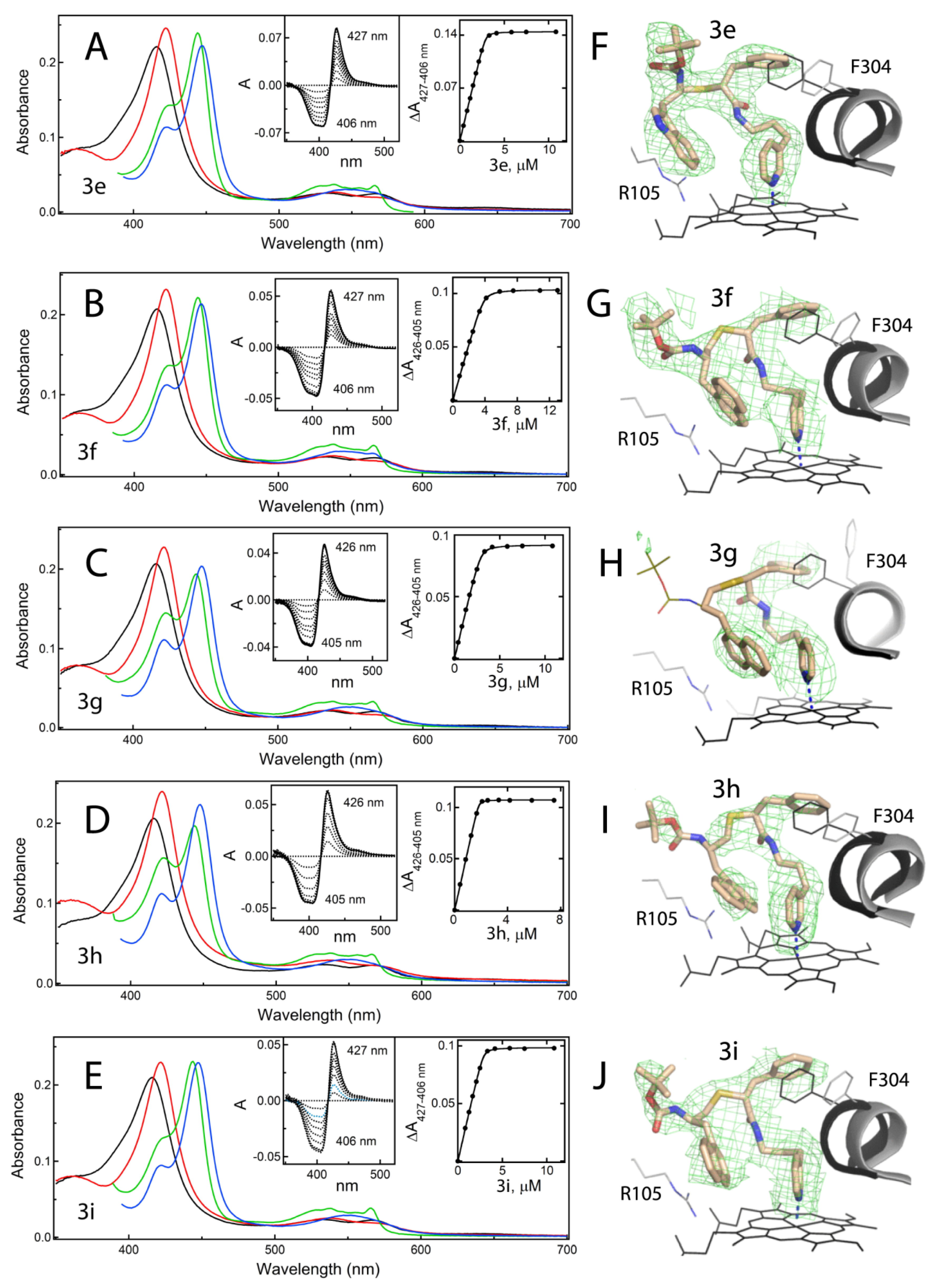
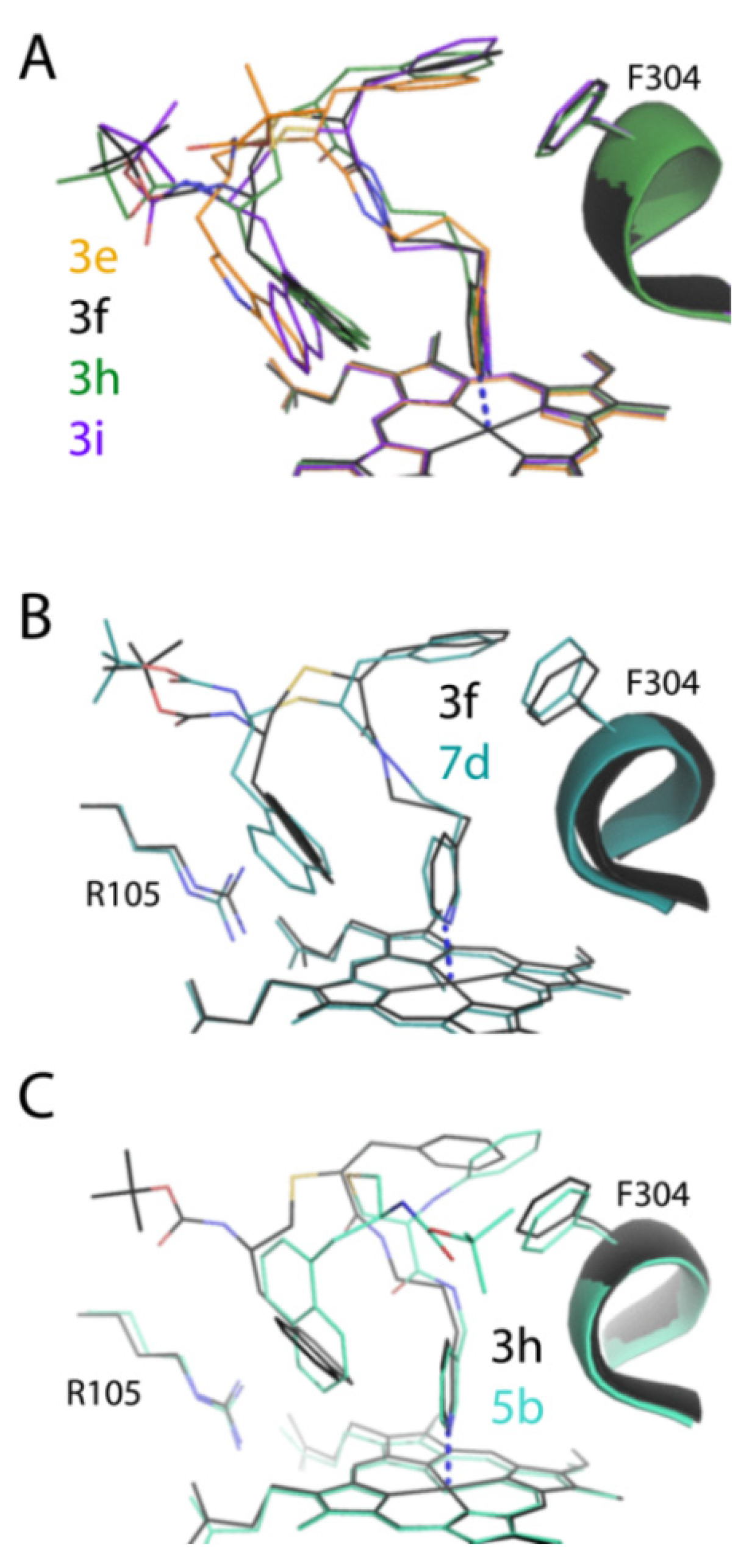
Another challenge was to accurately define the ligand binding modes. Despite the fact that most of the new compounds were the tight binders that willingly co-crystallized with CYP3A4, it was difficult to determine their binding orientation in traditional I222 crystals, containing one loosely packed CYP3A4 molecule in the unit cell. This and the higher motional freedom of elongated compounds led to thermal disorder and discontinuous electron density maps. To overcome this problem, we recrystallized some but not all inhibitory complexes in the more densely packed C2 and I2 space groups. As expected, changes in crystal packing triggered the restructuring of surface loops, including the F–F’ fragment (
Figure S1A–C), which nonetheless only minimally distorted the ligand binding modes (
Figure S2). Two compounds,
3d and
3i, failed to co-crystallize with WT and the previously obtained K282A/K285A CYP3A4 [
20] in the desired crystal forms. This prompted us to clone another surface mutant, K421A/K424A, which had similar spectral and ligand binding properties (
Figure S3), easily formed C2 crystals, and maintained the same structural fold (
Figure S1D). Thus, along with WT and K282A/K285A CYP3A4, this variant can be used in crystallization trials to increase chances for success.
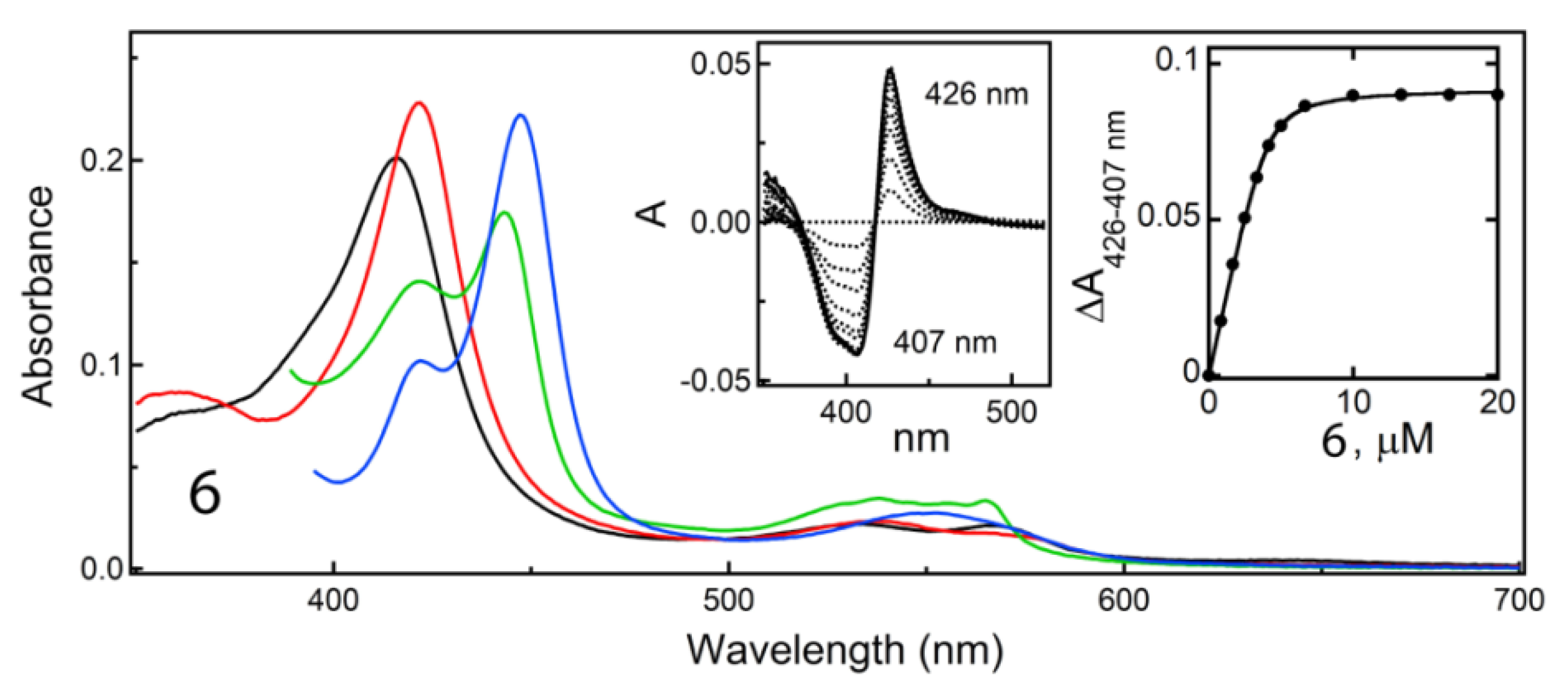
Determination of all but
6-bound structures of CYP3A4 was important because it enabled a structural comparison within/between subseries and with the pyridyl–ethyl counterparts (
Figure 4B and
Figure 7B,C). Comparative analysis suggests that the positive impact of one-atom linker extension on K
s and IC
50 could be due to the higher flexibility and adjustability of the elongated analogues, leading to stronger heme ligation and a more relaxed fit. This, in turn, strengthens the protein–ligand interactions mediated by the side-groups, as manifested by a large increase in thermostability of CYP3A4 (ΔT
m in
Table 1). There was also a downside effect, an increase in ligand mobility and thermal motion (evident from the elevated B-factors;
Tables S1 and S2), which could lower the inhibitory potency to some extent. This possibility explains why most series V analogues are inferior to the shorter
5b and
7d. Thus, a balance needs to be found between the flexibility/adjustability and rigidity of the scaffold. One way to achieve this would be through modification of the end-moiety, currently presented by a simple amino-protecting Boc-group. Due to a spacious active site and the adjacent substrate access channel (
Figure 9), the addition/optimization of the end-moiety might not only improve the inhibitory potency of the analogues, but also address the solubility and cross-reactivity issues associated with ritonavir.
Since the end-moiety has yet to be evaluated and optimized, it would be premature to draw any final conclusions on the rational inhibitor design. However, this study completes the investigation of the most important part of the ritonavir-like scaffold—the heme-ligating, side-group containing inhibitor core. A build-from-scratch approach utilized for inhibitor design, with systematic changing/testing of every pharmacophoric determinant, and the availability of structural information on the inhibitory complexes were critical for deciphering the relative importance of the structural elements of ritonavir. SAR analyses of the rationally designed analogues not only confirmed the early observations [
19], but also led to new discoveries and conclusions. In particular, all investigated compounds were found to cause the I-helix distortion, with the largest shift (>2.0 Å) observed in CYP3A4 complexed with strong binders/inhibitors, including ritonavir (
Figure 9). Such high plasticity of the I-helix enables the R
1–phenyl to insert deeper into P1 pocket, with no apparent penalty for the conformational constraint. However, a 2-fold drop in IC
50 observed upon the R
1–phenyl-to-naphthalene substitution [
10] suggests that the pocket has limited capacity to expand and can optimally accommodate groups no larger than phenyl. Importantly, hydrophobic and aromatic interactions established at the P1 site have a higher impact on K
s than IC
50. This was further demonstrated with the series V analogues, whose backbone elongation facilitated the insertion of R
1 (compare the R
1 insertion angle in
Figure 9).
In contrast, the R
2-mediated interactions at the P2 site largely affect the inhibitory potency, with naphthalene being the most optimal substituent. Notably, none of the designed analogues, including
3h and
7d, displace the I369-A370-M371 fragment as much as ritonavir: the A370 Cα atom shifts by 0.5, 0.9 and 1.8 Å, respectively (
Figure 9). Thus, the R
2-A370 clash is unfavourable and tends to be avoided or minimized. The active site expansion also proceeds through displacement of the F–F’ connecting loop, part of which (residues 210–214) is invisible in most of the CYP3A4 structures and does not seem to contribute much to the binding energy. Nonetheless, L210, L211 and F213 could establish hydrophobic interactions with the R
1–phenyl and Boc-group (
Figure S2B,D), which will be taken into consideration during optimization of the end-moiety.
In addition to the ability to strongly ligate to the heme via the N-pyridine heteroatom, all potent inhibitors establish an H-bond with the active site S119. The H-bonding propensity is diminished or completely lost in the S/R stereoisomers which, overall, have the lowest inhibitory potential. Stereochemistry becomes even more important when the side-groups are substituted with bulky indole and naphthalene, whose ability to adjust/rearrange in the active site is likely to be limited. This, in turn, could explain why the backbone flexibility and proper spacing between the functional groups are important and greatly improve the binding and inhibitory strength. Considering recent findings on P450 aromatase inhibitors [
43], it is plausible to suggest that strong heme ligation, favourable stereochemistry (when applicable), H-bonding to the active site residue(s), and the deformation of the active site (induced fit) might be common features leading to the potent inhibition of CYP enzymes.
In conclusion, the spectral, functional and structural characterization of eleven series V analogues showed that six- and seven-atom separation between the pyridine and R1 is most optimal, as further linker extension lowers the binding and inhibitory strength by several-fold, likely due to spatial limitations in the active site and/or conformational constraints. Compared to the shorter series IV analogues, an increase in the R2 size/hydrophobicity has a less pronounced impact, partly due to the higher flexibility/mobility of the elongated compounds. Even so, the new lead compound, 3h, was among the best CYP3A4 inhibitors designed to date with the highest binding affinity ever detected. Moreover, 3h and other lead compounds most effectively prevented the H2O2-induced degradation of the heme, confirming once again that strong heme ligation and the blockage of the active site are critical for potent inhibition. Finally, the SAR analysis of series IV and V analogues suggests that further improvement in the inhibitory power could be achieved by balancing the flexibility/adjustability and rigidity of the scaffold.
4. Materials and Methods
Chemistry General Methods—All reactions were performed with commercially available reagents (Aldrich, Thermo-Fisher, Alfa Aesar, Acros, Oakwood, Millipore) without further purification. Anhydrous solvents were acquired through a solvent purification system (Inert PureSolv and JC Meyer systems) or purified according to standard procedures.
1H NMR spectra were recorded on Bruker DRX 400 MHz, Bruker DRX 500 MHz, or Bruker Avance 600 MHz spectrometer and processed using TopSpin 3.5 software. Low and high resolution mass spectrometry (LRMS and HRMS) data were obtained via ESI LC-TOF on a Waters (Micromass) LCT Premier spectrometer (Waters), with PEG as the calibrant for HRMS. Optical rotation was recorded on a Rudolph Autopol III Automatic Polarimeter at room temperature in methanol. Thin layer chromatography (TLC) was performed using EMD Millipore silica gel 60 F
254 aluminium plates. Separation by column chromatography was conducted using Fisher silica gel 60 (230–400 mesh). All investigated compounds were >95% pure as determined by NMR. High resolution mass spectrometry data and NMR spectra are included in the
Supplementary Material.
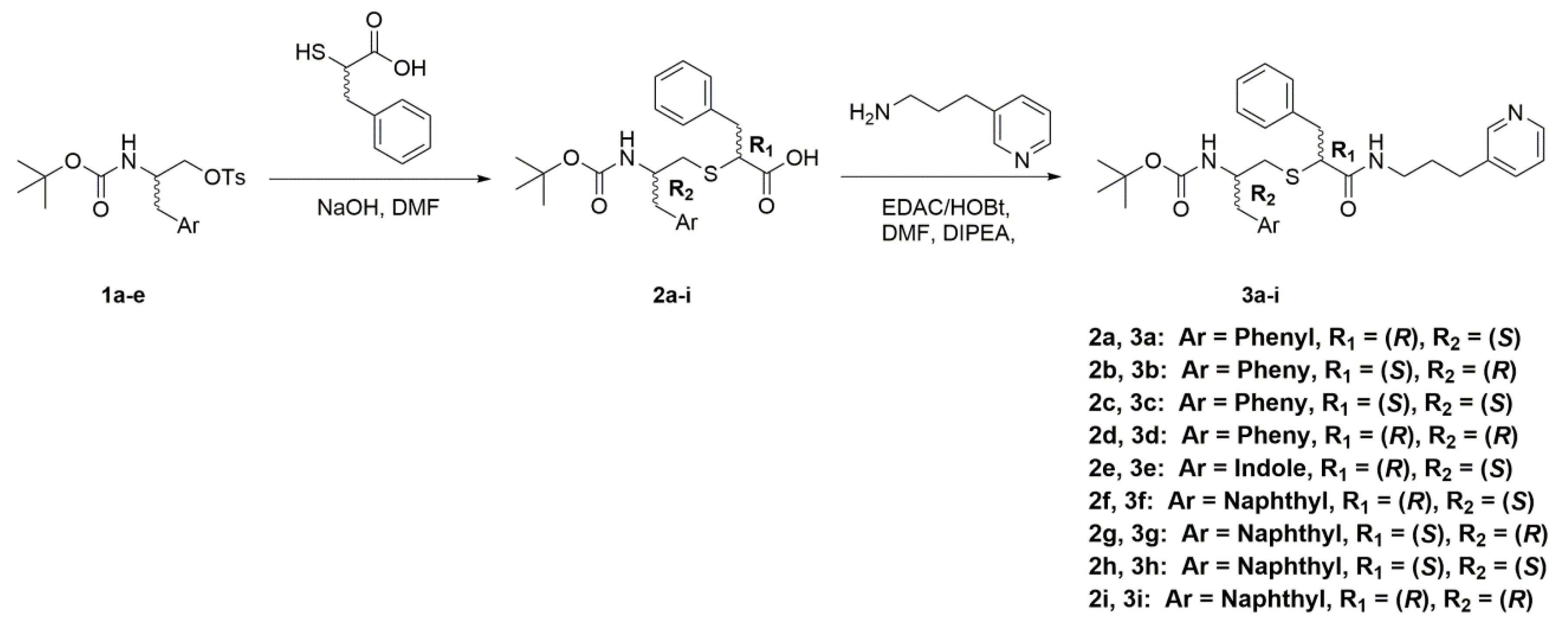
Synthesis of Analogues—Compounds
1a–
e were prepared as described previously [
10,
22], with either boc-protection and the tosylation of commercially available amino alcohol or amino acid reduction and protection. Sequentially, the Ar groups are:
1a phenyl (
S),
1b phenyl (
R),
1c indole (
S),
1d naphthyl (
S), and
1e naphthyl (
R). Compounds
2a–
i were also prepared as described previously [
10,
22], with either
D or
L-α-thio-phenylalanine [
44]. Synthesis of other analogues is outlined in
Scheme 1,
Scheme 2 and
Scheme 3.
General Procedure for the Synthesis of Compounds
3a–
i— Steps for the synthesis of compounds
3a-
i are outlined in
Scheme 1. Crude
2a (0.34 g, 0.82 mmol) was dissolved in dimethylformamide (DMF) (3 mL). To this solution, 3-(3-pyridyl)propylamine (0.17 g, 1.23 mmol, 1.5 eq) in 3 mL DMF was added, followed by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) (0.24 g, 1.23 mmol, 1.5 eq), hydroxybenzotriazole (HOBt) (0.19 g, 1.23 mmol, 1.5 eq) and N,N-diisopropylethylamine (DIPEA) (0.32 g, 2.46 mmol, 3 eq). The reaction was stirred at room temperature overnight. Upon completion, the solvent was evaporated and the reaction mixture was diluted with ethyl acetate. The organic layer was then washed with saturated NaHCO
3, water, and brine. The combined organic layers were dried over MgSO
4 and concentrated in vacuo to give the crude product, which was purified via column chromatography (95:5 EtOAc:MeOH). The pure product
3a was obtained as a clear yellow oil (0.07 g, 16%). TLC: EtOAc/MeOH 90:10 (Rf. 0.57).
1H NMR (400 MHz, CDCl
3) δ 8.43 (d,
J = 4.8 Hz, 1H), 8.37 (s, 1H), 7.43 (d,
J = 7.7 Hz, 1H), 7.36–7.11 (m, 11H), 6.52 (t,
J = 6.0 Hz, 1H (NH)), 4.63 (d,
J = 8.3 Hz, 1H), 3.96 (m, 1H), 3.25 (quint,
J = 6.7 Hz, 2H), 2.90 (m, 2H), 2.79 (d,
J = 6.9 Hz, 2H), 2.68 (dd,
J = 5.2, 13.6 Hz, 1H), 2.58 (dd,
J = 6.1, 13.5 Hz, 1H), 2.48 (t,
J = 7.9 Hz, 2H), 1.69 (quint,
J = 7.3 Hz, 2H), 1.40 (s, 9H). HRMS
m/z calculated for C
31H
39N
3O
3SNa [M + Na]
+: 556.2610. Found: 556.2606. The pure product
3b was obtained as a yellow gum (0.015 g, 15%). TLC: EtOAc/MeOH 90:10 (Rf. 0.57).
1H NMR (400 MHz, CDCl
3) δ 8.45 (d,
J = 4.9 Hz, 1H), 8.39 (s, 1H), 7.44 (d,
J = 8.1 Hz, 1H), 7.31–7.16 (m, 9H), 7.13 (d,
J = 6.8 Hz, 2H), 6.45 (t,
J = 7.0 Hz, 1H (NH)), 4.57 (bs, 1H), 3.96 (m, 1H), 3.26 (quint,
J = 7.0 Hz, 2H), 3.18 (m, 1H), 2.96 (dd,
J = 7.0, 13.4 Hz, 1H), 2.79 (d,
J = 7.0 Hz, 2H), 2.67 (dd,
J = 5.7 13.5 Hz, 1H), 2.58 (dd,
J = 6.0, 13.4 Hz, 1H), 2.49 (t,
J = 7.7 Hz, 2H), 1.70 (quint,
J = 7.4 Hz, 2H), 1.41 (s, 9H). HRMS
m/z calculated for C
31H
39N
3O
3SNa [M + Na]
+: 556.2610. Found: 556.2587. The pure product
3c was obtained as a yellow gum (0.018 g, 18%). TLC: EtOAc/MeOH 90:10 (Rf. 0.57).
1H NMR (400 MHz, CDCl
3) δ 8.44 (d,
J = 4.8 Hz, 1H), 8.39 (s, 1H), 7.45 (d,
J = 7.6 Hz, 1H), 7.32–7.17 (m, 9H), 7.08 (d,
J = 6.7 Hz, 2H), 6.90 (bs, 1H (NH)), 4.64 (d,
J = 6.9 Hz, 1H), 3.85 (q,
J = 6.7 Hz, 1H), 3.54 (m, 1H), 3.30 (dd,
J = 7.4, 13.7 Hz, 2H), 3.19 (quint,
J = 6.7 Hz, 1H), 2.91 (m, 2H), 2.68 (m, 2H), 2.50 (m, 2H), 1.73 (quint,
J = 7.5 Hz, 2H), 1.37 (s, 9H). HRMS
m/z calculated for C
31H
39N
3O
3SNa [M + Na]
+: 556.2610. Found: 556.2585. The pure product
3d was obtained as a yellow gum (0.027 g, 21%). TLC: EtOAc/MeOH 90:10 (Rf. 0.57).
1H NMR (400 MHz, CDCl
3) δ
1H NMR (400 MHz, CDCl
3) δ 8.44 (d,
J = 4.8 Hz, 1H), 8.37 (s, 1H), 7.45 (d,
J = 7.8 Hz, 1H), 7.31–7.17 (m, 9H), 7.08 (d,
J = 6.8 Hz, 2H), 6.89 (bs, 1H (NH)), 4.64 (d,
J = 7.8 Hz, 1H), 3.85 (q,
J = 7.0 Hz, 1H), 3.54 (m, 1H), 3.30 (dd,
J = 7.3, 13.7 Hz, 2H), 3.19 (quint,
J = 6.7 Hz, 1H), 2.91 (m, 2H), 2.69 (m, 2H), 2.49 (m, 2H), 1.73 (quint,
J = 7.4 Hz, 2H), 1.39 (s, 9H). HRMS
m/z calculated for C
31H
39N
3O
3SNa [M + Na]
+: 556.2610. Found: 556.2598. Compound
3e was obtained as a white fluffy solid (0.022 g, 13%). TLC: EtOAc/MeOH 90:10 (Rf. 0.49).
1H NMR (400 MHz, CDCl
3) δ 8.44 (d,
J = 4.8 Hz, 1H), 8.33 (s, 1H). In addition, 8.30 (s, 1H), 7.61 (d,
J = 7.6 Hz, 1H), 7.41 (d,
J = 7.8 Hz, 1H), 7.38–7.13 (m, 7H), 7.10 (t,
J = 6.9 Hz, 1H), 6.98 (s, 1H), 6.33 (t,
J = 5.2 Hz, 1H (NH)), 4.69 (d,
J = 7.7 Hz, 1H), 4.09 (q,
J = 6.6 Hz, 1H), 3.25 (dd,
J = 7.4, 13.7 Hz, 1H), 3.12 (quint,
J = 6.8 Hz, 1H), 2.97 (m, 4H), 2.71 (dd,
J = 5.5, 13.9 Hz, 1H), 2.59 (dd,
J = 6.2, 13.4 Hz, 1H), 2.40 (t,
J = 7.7 Hz, 2H), 1.56 (quint,
J = 7.4 Hz, 2H), 1.43 (s, 9H). HRMS
m/z calculated for C
33H
41N
4O
3S [M + H]
+: 573.2899. Found: 573.2900. The pure product
3f was obtained as an off white fluffy solid (0.031 g, 20%). TLC: EtOAc/MeOH 90:10 (Rf. 0.51).
1H NMR (400 MHz, CDCl
3) δ 8.43 (d,
J = 4.7 Hz, 1H), 8.13 (d,
J = 8.4 Hz, 1H), 7.82 (d,
J = 8.0 Hz, 1H), 7.72 (d,
J = 8.1 Hz, 1H), 7.55 (t,
J = 7.8 Hz, 1H), 7.47 (t,
J = 8.0 Hz, 1H), 7.36 (m, 3H), 7.31–7.16 (m, 7H), 6.34 (t,
J = 5.3 Hz, 1H (NH)), 4.73 (d,
J = 7.9 Hz, 1H), 4.16 (q,
J = 6.3 Hz, 1H), 3.25 (m, 4H), 2.96 (dd,
J = 6.8, 13.7 Hz, 2H), 2.68 (t,
J = 4.6 Hz, 2H), 2.36 (t,
J = 7.8 Hz, 2H), 1.53 (t,
J = 7.4 Hz, 2H), 1.39 (s, 9H). HRMS
m/z calculated for C
35H
42N
3O
3S [M + H]
+: 584.2947. Found: 584.2921. The pure product
3g was obtained as a yellow solid (0.024 g, 16%). TLC: EtOAc/MeOH 90:10 (Rf. 0.5).
1H NMR (400 MHz, CDCl
3) δ 8.44 (d,
J = 4.7 Hz, 1H), 8.13 (d,
J = 8.3 Hz, 1H), 7.82 (d,
J = 8.2 Hz, 1H), 7.72 (d,
J = 8.0 Hz, 1H), 7.55 (t,
J = 7.6 Hz, 1H), 7.47 (t,
J = 8.0 Hz, 1H), 7.36 (m, 3H), 7.31–7.16 (m, 7H), 6.32 (t,
J = 6.8 Hz, 1H (NH)), 4.72 (d,
J = 8.0 Hz, 1H), 4.16 (q,
J = 6.5 Hz, 1H), 3.25 (m, 4H), 2.96 (dd,
J = 6.8, 13.7 Hz, 2H), 2.68 (t,
J = 4.8 Hz, 2H), 2.36 (t,
J = 6.8 Hz, 2H), 1.54 (t,
J = 6.7 Hz, 2H), 1.39 (s, 9H). HRMS
m/z calculated for C
35H
42N
3O
3S [M + H]
+: 584.2947. Found: 584.2930. The pure product
3h was obtained as an off white fluffy solid (0.089 g, 26%). TLC: EtOAc/MeOH 90:10 (Rf. 0.51 (Avg)).
1H NMR (400 MHz, CDCl
3) δ 8.43 (d,
J = 4.8 Hz, 1H), 8.35 (s, 1H), 8.16 (d,
J = 7.8 Hz, 1H), 7.88 (d,
J = 7.6 Hz, 1H), 7.77 (d,
J = 8.2 Hz, 1H), 7.53 (quint,
J = 7.6 Hz, 2H), 7.40 (t,
J = 7.5 Hz, 2H), 7.29–7.16 (m, 7H), 6.81 (t,
J = 5.3 Hz, 1H (NH)), 4.77 (d,
J = 7.1 Hz, 1H), 4.08 (q,
J = 6.4 Hz, 1H), 3.53 (m, 1H), 3.30 (m, 2H), 3.11 (m, 2H), 2.93 (dd,
J = 6.0, 13.6 Hz, 1H), 2.79 (m, 1H), 2.54 (dd,
J = 6.6, 13.9 Hz, 1H), 2.43 (sext,
J = 6.3 Hz, 2H), 1.67 (oct,
J = 7.4 Hz, 2H), 1.34 (s, 9H). HRMS
m/z calculated for C
35H
42N
3O
3S [M + H]
+: 584.2947. Found: 584.2928. The pure product
3i was obtained as a white fluffy solid (0.07 g, 26%). TLC: EtOAc/MeOH 90:10 (Rf. 0.49).
1H NMR (400 MHz, CDCl
3) δ 8.43 (d,
J = 4.7 Hz, 1H), 8.35 (s, 1H), 8.16 (d,
J = 7.8 Hz, 1H), 7.86 (dd,
J = 8.2, 16.5 Hz, 1H), 7.75 (dd,
J = 8.0, 19.1 Hz, 1H), 7.53 (m,
J = 7.7 Hz, 2H), 7.40 (t,
J = 7.7 Hz, 2H), 7.30–7.15 (m, 7H), 6.82 (bs, 1H (NH)), 4.77 (d,
J = 8.0 Hz, 1H), 4.08 (sext,
J = 6.7 Hz, 1H), 3.53 (m, 1H), 3.31 (dd,
J = 8.5, 13.5 Hz, 2H), 3.13 (dd,
J = 8.0, 14.8 Hz, 2H), 2.93 (dd,
J = 6.2, 13.5 Hz, 1H), 2.79 (m, 1H), 2.54 (dd,
J = 6.9, 14.3 Hz, 1H), 2.43 (sext,
J = 5.9 Hz, 2H), 1.67 (sext,
J = 7.6 Hz, 2H), 1.34 (s, 9H). HRMS
m/z calculated for C
35H
41N
3O
3SNa [M + Na]
+: 606.2766. Found: 606.2797.
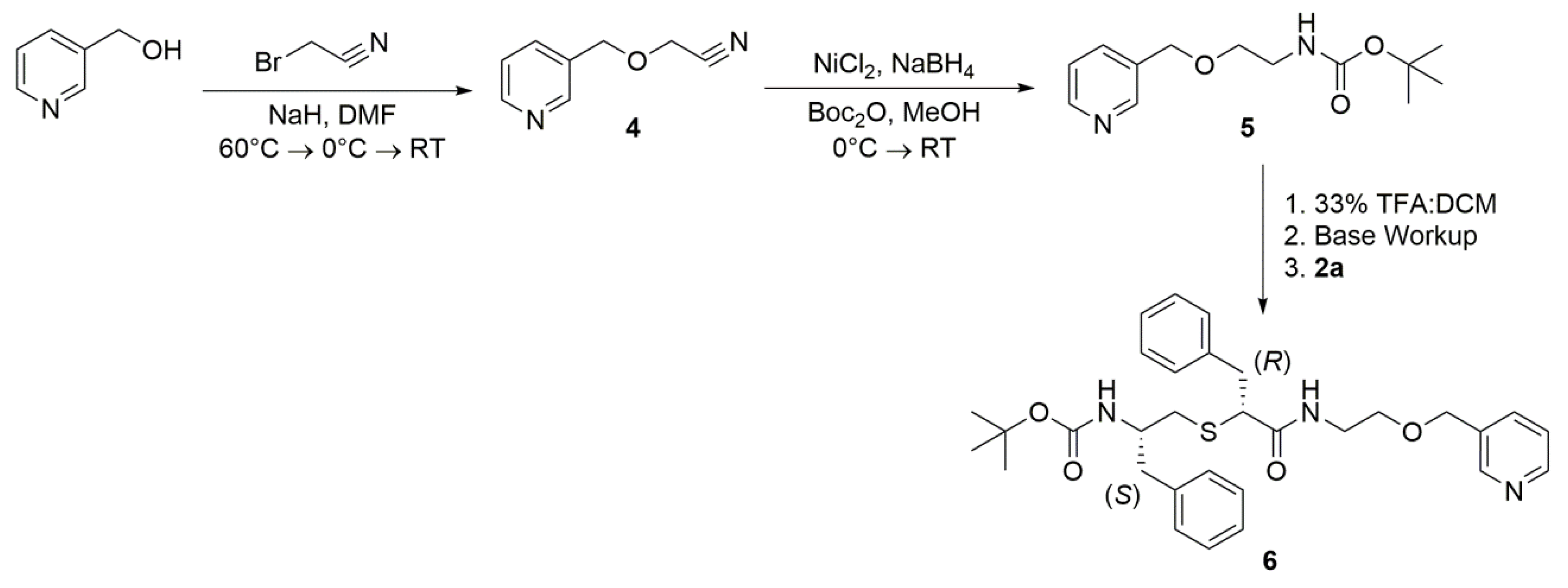
Synthesis of Compound 4—Pyridine-3-methanol (2.0 g, 18.3 mmol) was dissolved in anhydrous DMF (30 mL) and the solution was cooled to 0 °C on an ice bath. Sodium hydride (0.88g, 36.6 mmol, 2 eq) was added slowly over 10 min. Once H2 formation subsided, the solution was warmed to 60 °C and stirred for 45 min. The solution was then cooled to 0 °C on an ice bath and bromoacetonitrile (2.74 g, 22.8 mmol, 1.25 eq) was added dropwise. The reaction was allowed to slowly come to room temperature overnight. Upon completion, the solvent was evaporated, and the reaction mixture was diluted with ethyl acetate. The organic layer was then washed with saturated NaHCO3, water, and brine. The combined organic layers were dried over MgSO4, filtered through a silica plug, and concentrated in vacuo to give the crude product, which was purified via column chromatography (100% EtOAc). The pure product 4 was obtained as a brown oil (0.65g, 24%). LRMS m/z calculated for C8H8N2ONa [M + Na]+: 171.0. Found: 171.0
Synthesis of Compound 5—Compound 4 (0.5 g, 3.4 mmol) was dissolved in methanol (20 mL) and the solution was cooled to 0 °C on an ice bath. Nickel chloride hexahydrate (0.89 g, 3.7 mmol, 1.1 eq,) and Di-tert-butyl dicarbonate (1.5 g, 6.8 mmol, 2 eq) were then added to the solution, followed by slow addition of sodium borohydride (1.3 g, 34 mmol, 10 eq) over 10 min. Once H2 formation subsided, the solution was removed from the ice bath and allowed to stir at room temperature overnight. Upon completion, the solvent was evaporated, and the reaction mixture was diluted with ethyl acetate. The organic layer was then washed with saturated NaHCO3 and filtered through celite. The organic layers were dried over MgSO4 and concentrated in vacuo to give the crude product, which was purified via column chromatography (100% EtOAc). The pure product 5 was obtained as a brown oil (0.1 g, 12%). LRMS m/z calculated for C13H20N2O3Na [M + Na]+: 275.1. Found: 275.1.
Synthesis of Compound 6—First, compound 5 (0.1 g, 0.40 mmol) was added to dichloromethane (DCM) (3 mL). Trifluoroacetic acid (1.5 mL) was added dropwise and the reaction was allowed to stir at room temperature for 2 h. Upon completion, the solvent was evaporated, and the reaction mixture was neutralized with NaHCO3. The pH was then adjusted to 14 with NaOH, and the solution was extracted with DCM. The combined organic layers were concentrated in vacuo to give the free amine product (2-(pyridine-3-ylmethoxy)ethan-1-amine) as a light-yellow oil (0.06g, 98%). HRMS m/z calculated for C8H13N2O [M + H]+: 153.1028. Found: 153.1043. Then, crude 2a (0.15 g, 0.36 mmol) was dissolved in DMF (3 mL). To this solution, EDAC (0.1 g, 0.54 mmol, 1.5 eq) and HOBt (0.083 g, 0.54 mmol, 1.5 eq) were added, followed by the addition of 2-(pyridine-3-ylmethoxy)ethan-1-amine (0.06 g, 0.39 mmol, 1.1 eq) in DMF (2 mL) and DIPEA (0.14 g, 1.1 mmol, 3 eq). The reaction was stirred at room temperature overnight. Upon completion, the solvent was evaporated, and the reaction mixture was diluted with ethyl acetate. The organic layer was then washed with saturated NaHCO3, water, and brine. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give the crude product, which was purified via column chromatography (100% EtOAc). The pure product 6 was obtained as a light orange oil (0.013 g, 6.5%). TLC: EtOAc/MeOH 90:10 (Rf. 0.54). 1H NMR (400 MHz, CDCl3) δ 8.54 (m, 2H), 7.64 (d, J = 6.2 Hz, 1H), 7.34–7.07 (m, 11H), 6.75 (m, 1H (NH)), 4.43 (s, 1H), 3.64 (m, 2H), 3.47 (m, 2H), 3.36 (s, 1H), 3.25 (dd, J = 6.9, 13.4 Hz, 1H), 2.95 (m, 2H), 2.73 (m, 2H), 2.65 (d, J = 6.8, 1H), 2.54 (dd, J = 6.5, 13.9 Hz, 1H), 2.35 (dd, J = 4.5, 19.4 Hz, 1H), 1.41 (s, 9H). HRMS m/z calculated for C31H40N3O4S [M + H]+: 550.2739. Found: 550.2738.
Compound
7 was prepared as described previously [
21], with n-phenylcysteine and Boc-protected, tosylated L-phenylalanine (Boc–L–Phe–OTs).
Synthesis of Compound 8—Crude 7 (0.34 g, 0.8 mmol) was dissolved in DMF (9 mL). To this solution, EDAC (0.23 g, 1.2 mmol, 1.5 eq) and HOBt (0.18 g, 1.2 mmol, 1.5 eq) were added, followed by the addition of 3-(3-pyridyl)propylamine (0.16 g, 1.2 mmol, 1.5 eq) and DIPEA (0.31 g, 2.4 mmol, 3 eq). The reaction was stirred at room temperature overnight. Upon completion, the solvent was evaporated, and the reaction mixture was diluted with ethyl acetate. The organic layer was then washed with saturated NaHCO3, water, and brine. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give the crude product, which was purified via column chromatography (95:5 EtOAc:MeOH). The pure product 8 was acquired as a light-yellow gum (0.032 g, 23%). TLC: EtOAc/MeOH 90:10 (Rf. 0.55). 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 4.7 Hz, 1H), 8.36 (s, 1H), 7.41 (m, 2H), 7.30–7.12 (m, 6H), 7.00 (t, J = 6.1 Hz, 1H), 6.85 (dt, J = 3.4, 11.0 Hz, 1H), 6.67 (t, J = 8.5 Hz, 2H), 4.70 (t, J = 10.6 Hz, 1H), 4.61 (bs, 1H (NH)), 4.00 (bs, 1H), 3.86 (bd, J = 28.8 Hz, 1H (NH)), 3.30 (m, 2H), 3.15 (m, 1H), 3.00 (m, 1H), 2.85 (m, 2H), 2.64 (m, 2H), 2.56 (t, J = 7.7 Hz, 2H), 1.87 (bs, 1H (NH)), 1.78 (m, 2H), 1.42 (d, J = 3.0 Hz, 9H). HRMS m/z calculated for C31H40N4O3SNa [M + Na]+: 571.2719. Found: 571.2698.
Protein Expression and Purification—Codon-optimized full-length and Δ3–22 human CYP3A4 were produced as reported previously [
45] and used for assays and crystallization, respectively. The K421A and K424A mutations were introduced to the Δ3–22 CYP3A4 expression plasmid using a QuikChange mutagenesis kit (Stratagene) and confirmed by sequencing. The mutant was expressed and purified similar to the wild-ype (WT) protein.
Spectral Binding Titrations—Equilibrium ligand binding to CYP3A4 was monitored in a Cary 300 spectrophotometer at ambient temperature in 0.1 M phosphate buffer, pH 7.4, supplemented with 20% glycerol and 1 mM dithiothreitol. Inhibitors were dissolved in dimethyl sulfoxide (DMSO) and added to a 2 μM protein solution in small aliquots, with the final solvent concentration < 2%. Spectral dissociation constants (Ks) were determined from quadratic fits to titration plots.
Thermal Denaturation—Thermal denaturation curves were recorded in 0.1 M phosphate buffer, pH 7.4, in a Cary 300 spectrophotometer. Protein (1 μM) was mixed with a ligand (20 μM) and incubated for 15 min at 23 °C. Melting curves were recorded at 260 nm using a 0.2 °C measurement step, 0.9 °C/min ramp rate, and a 50–75 °C temperature range. A denaturation midpoint (melting temperature; T
m) was determined from non-linear fittings to the melting curve as described earlier [
22].
Inhibitory Potency Assays—Inhibitory potency for the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC)
O-debenzylation activity of CYP3A4 was evaluated fluorometrically in a soluble reconstituted system. The full-length CYP3A4 and rat cytochrome P450 reductase (40 and 60 μM, respectively) were preincubated at room temperature for 1 h before 20-fold dilution with the reaction buffer consisting of 0.1 M potassium phosphate, pH 7.4, catalase and superoxide dismutase (2 units/mL each), and 0.0025% CHAPS (3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate). Prior to measurements, 85 μL of the reaction buffer was mixed with 10 μL of the NADPH-regenerating system (10 mM glucose, 0.2 mM NADP
+, and 2 units/mL glucose-6-phosphate dehydrogenase), 5 μL of the protein mixture (0.1 μM final CYP3A4 concentration), and 2 μL of the inhibitor solution or DMSO. The mixture was incubated for 2 min, after which 20 μL BFC and 70 μL NADPH (final concentration) were added to initiate the reaction. Accumulation of the fluorescent product, 7-hydroxy-4-(trifluoromethyl)coumarin, was monitored for 2 min at room temperature in a Hitachi F400 fluorimeter (λ
ex = 404 nm; λ
em = 500 nm). Within this time interval, fluorescence changes were linear. The average of three measurements was used to calculate the remaining activity, with the DMSO-containing sample used as a control (100% activity). The IC
50 values were derived from the (% activity) vs. (inhibitor concentration) plots as described previously [
22]. Our prior work showed that preincubation of ritonavir or the analogues with NADPH in a lipid-free reconstituted system leads to a small increase rather than decrease in IC
50, likely to due to partial metabolism [
22]. Therefore, in this study, inhibitory assays with NADPH preincubation were not conducted.
Kinetics of Ligand Binding and Heme Reduction—Kinetics of ligand binding to CYP3A4 and its reduction with sodium dithionite were measured at 426 and 443 nm, respectively, in a SX.18MV stopped flow apparatus (Applied Photophysics, UK), as described earlier [
22].
H2O2-Dependent Heme Depletion Assay—Heme bleaching in ligand-free and inhibitor-bound CYP3A4 (1.6 μM) was monitored at ambient temperature in 0.1 M phosphate buffer, pH 7.4. After 10 min preincubation of CYP3A4 with 16 μM inhibitors, 10 mM H2O2 (final concentration) was added, and heme decay was monitored at 420 nm for 120 min. The percentage of heme destroyed at the end of the reaction was calculated relative to that in the ligand-free CYP3A4 (100% decay).
Crystallization of the Inhibitory Complexes—Compounds 3a–d, 8, and 3e–h were co-crystallized with WTΔ3-22 CYP3A4, and 3d and 3i with the K421A/K424A mutant. Crystals of 3b-, 3c-, 3f-, 3g- and 8-bound complexes were grown using a microbatch method under paraffin oil. Other compounds were co-crystallized with CYP3A4 using a sitting drop vapor diffusion method. Prior to crystallization setup, CYP3A4 (60–70 mg/mL in 20–100 mM phosphate, pH 7.4) was incubated with a 2-fold ligand excess and centrifuged to remove the precipitate. The supernatant (0.4–0.6 μL) was mixed with 0.4–0.6 μL of crystallization solution containing: 8%–10% polyethylene glycol (PEG) 3350 and 70–80 mM sodium malonate, pH 7.0, for 3a, 3g and 8; 10%–12% PEG 3350 and 60–100 mM succinate, pH 7.0, for 3b and 3f; 10% PEG 3350 and 80 mM taximate, pH 6.0 (Hampton Research) for 3c; 6% PEG 3350 and 50 mM sodium malonate, pH 7.0, for 3e; or 6%–8% PEG 4000, 50 mM HEPES, pH 8.0, and 0.1–0.2 M lithium sulfate for 3d, 3h and 3i. Crystals were grown at room temperature for 2–3 days and cryoprotected with Paratone-N before freezing in liquid nitrogen.
Determination of the X-Ray Structures—X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource beamlines 9–2 and 12–2, and the Advanced Light Source beamlines 5.0.2 and 8.2.2. Crystal structures were solved by molecular replacement with PHASER [
46]. In addition, 5VCC was used as a search model for the
3b-,
3c-,
3f-,
3g- and
8-bound complexes, crystallized in the I222 space group. For the
3a-,
3d-,
3h- and
3i-bound CYP3A4, crystallized in the C2 and I2 space groups, the search model was the CYP3A4-
3e dimer, whose structure was determined first using 6UNJ for molecular replacement. Ligands were built with eLBOW [
47] and manually fit into the density with COOT [
48]. The initial models were rebuilt and refined with COOT and PHENIX [
47]. For racemic
8, the R
1 side-group configuration was automatically assigned to
R during the first refinement cycle. The following refinement was conducted with all stereoisomer combinations to confirm that the assigned chirality was most optimal. Polder omit electron density maps and local correlation coefficients (
Table S3) were calculated with PHENIX. Data collection and refinement statistics are summarized in
Tables S1 and S2. The atomic coordinates and structure factors for the
3a-,
3b-,
3c-,
3d-,
8-,
3e-,
3f-,
3g-,
3h-, and
3i-bound CYP3A4 were deposited in the Protein Data Bank with the ID codes 7KVH, 7KVI, 7KVJ, 7KVK, 7KVM, 7KVN, 7KVO, 7KVP, 7KVQ and 7KVS, respectively.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}