
Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
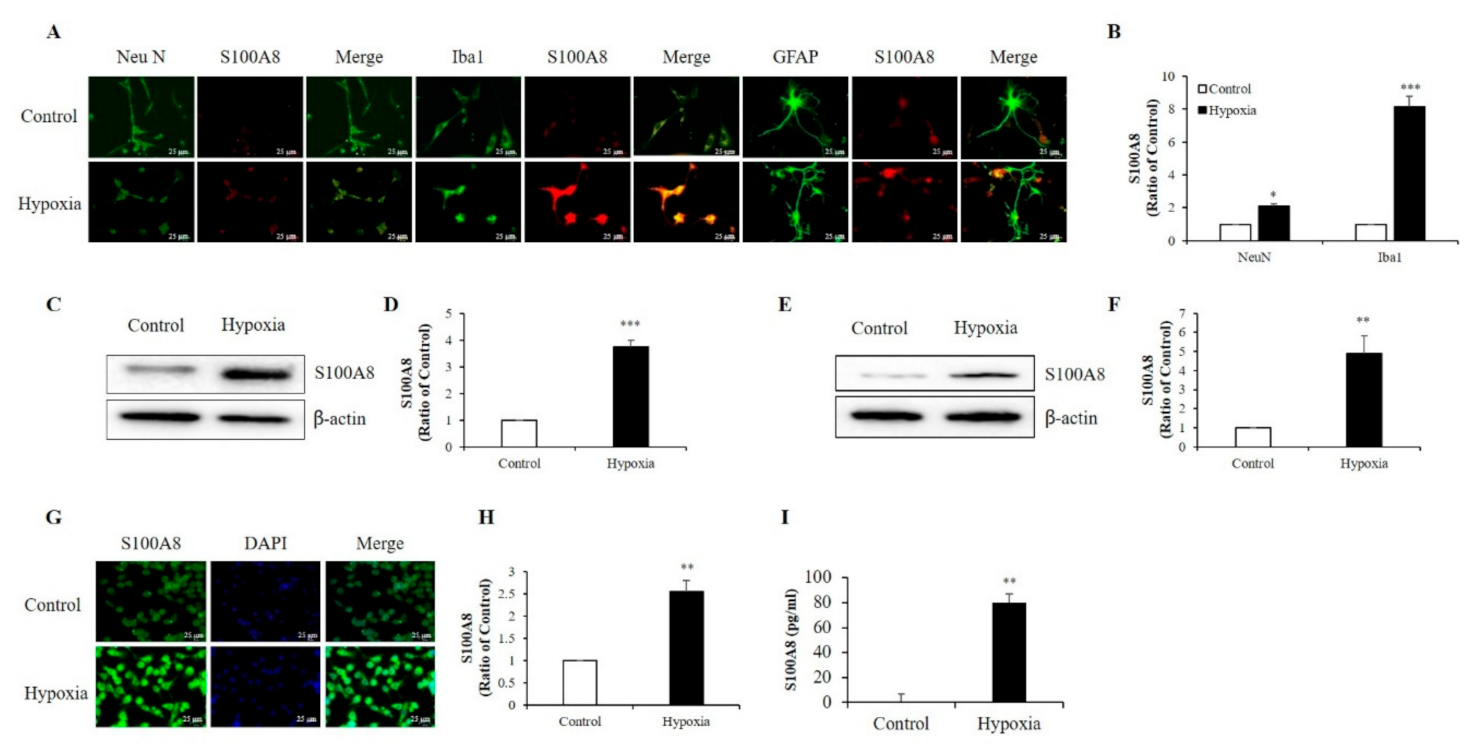
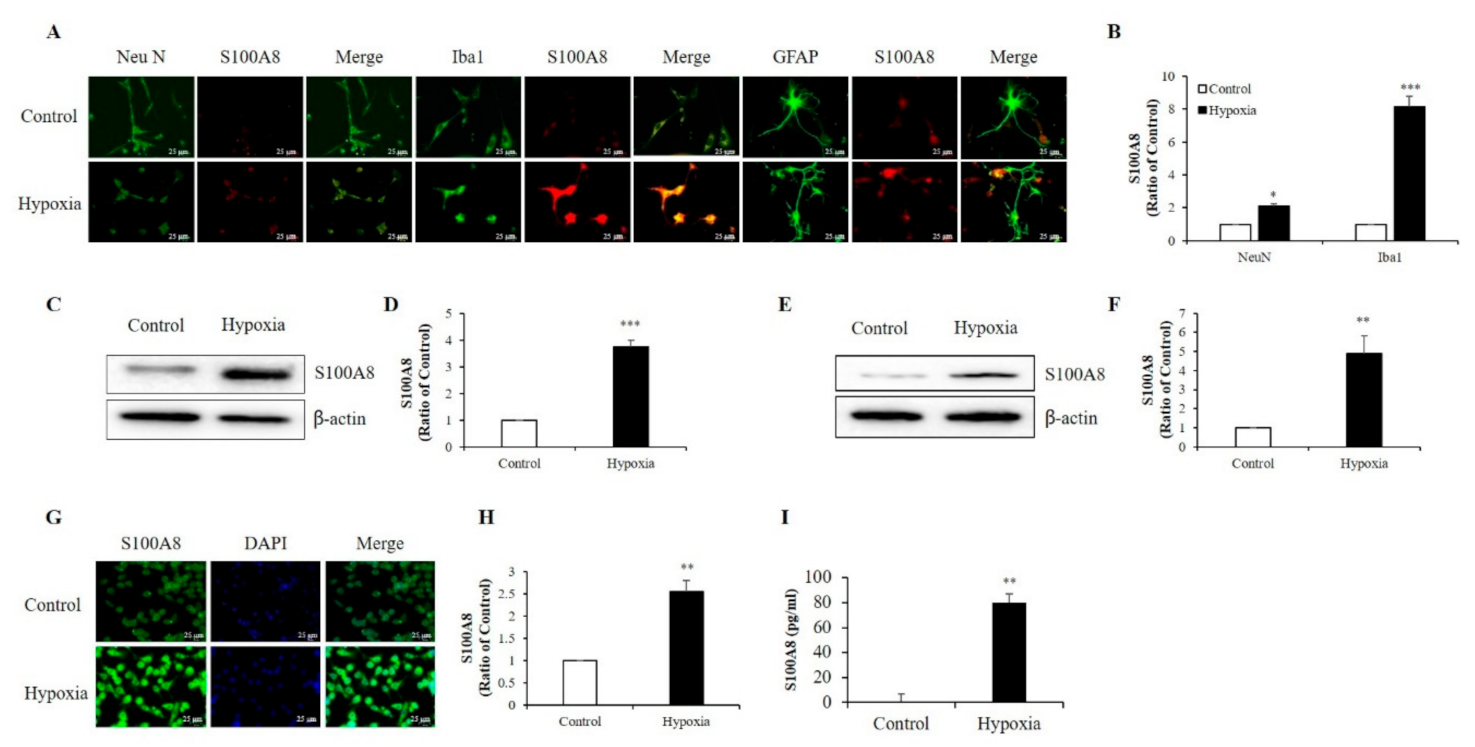
2.1. Hypoxia Increased the Production of S100A8 Protein in Neuronal and Microglial Cells and Induced the Secretion of S100A8 in SH-SY5Y Cells
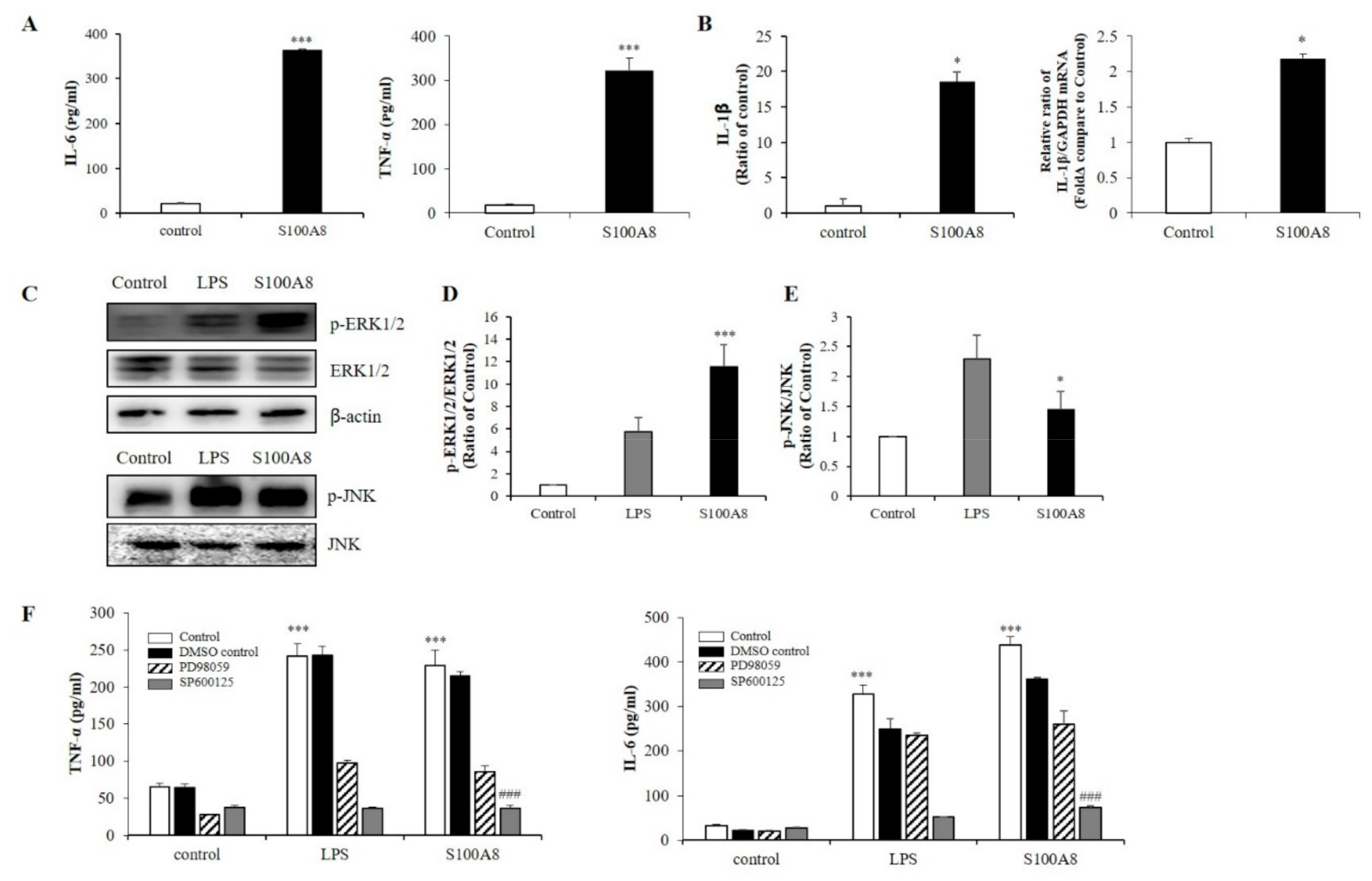
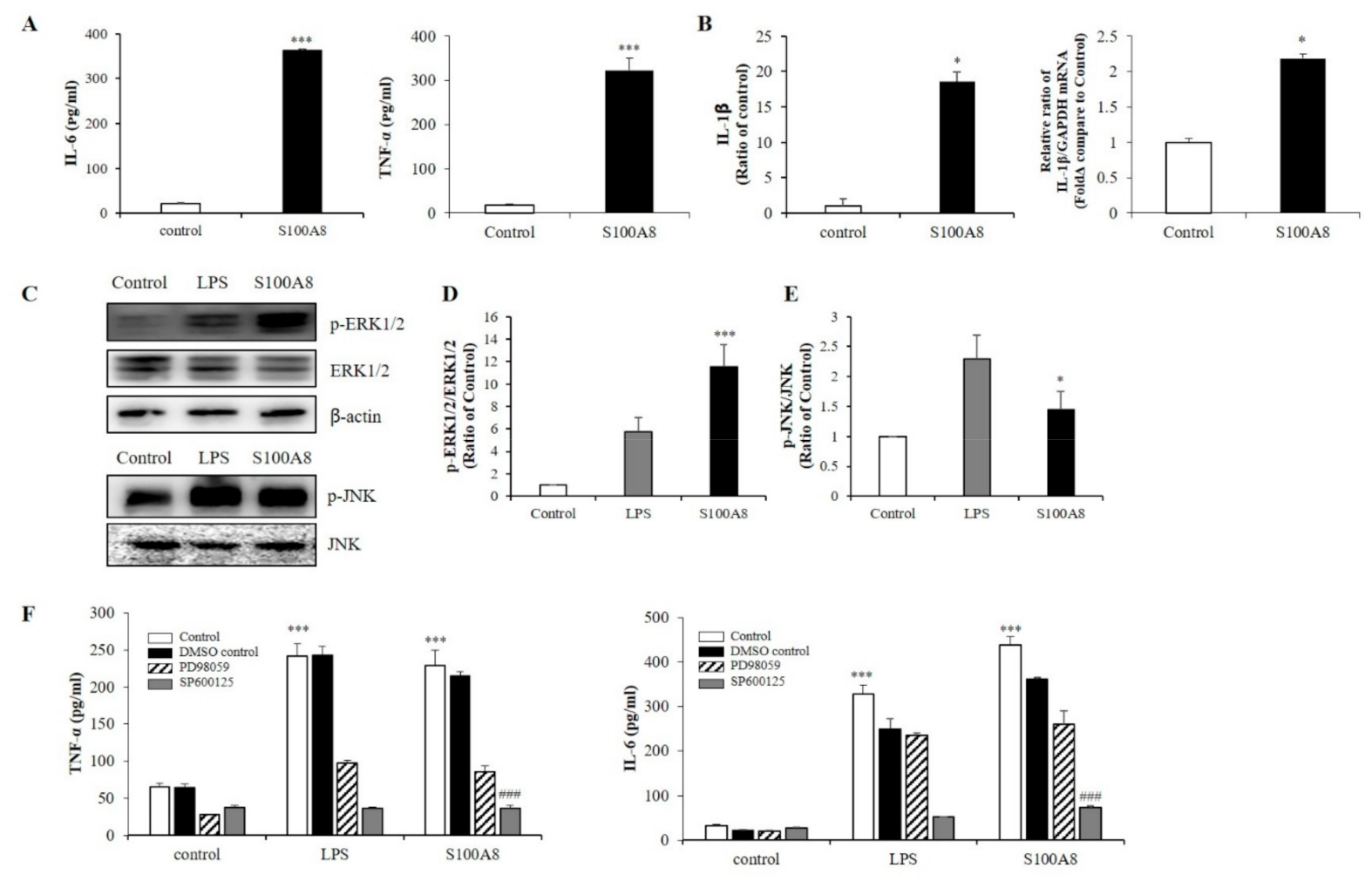
2.2. S100A8 Induced Pro-Inflammatory Cytokine Production Via Phosphorylation of ERK and JNK in BV-2 Cells
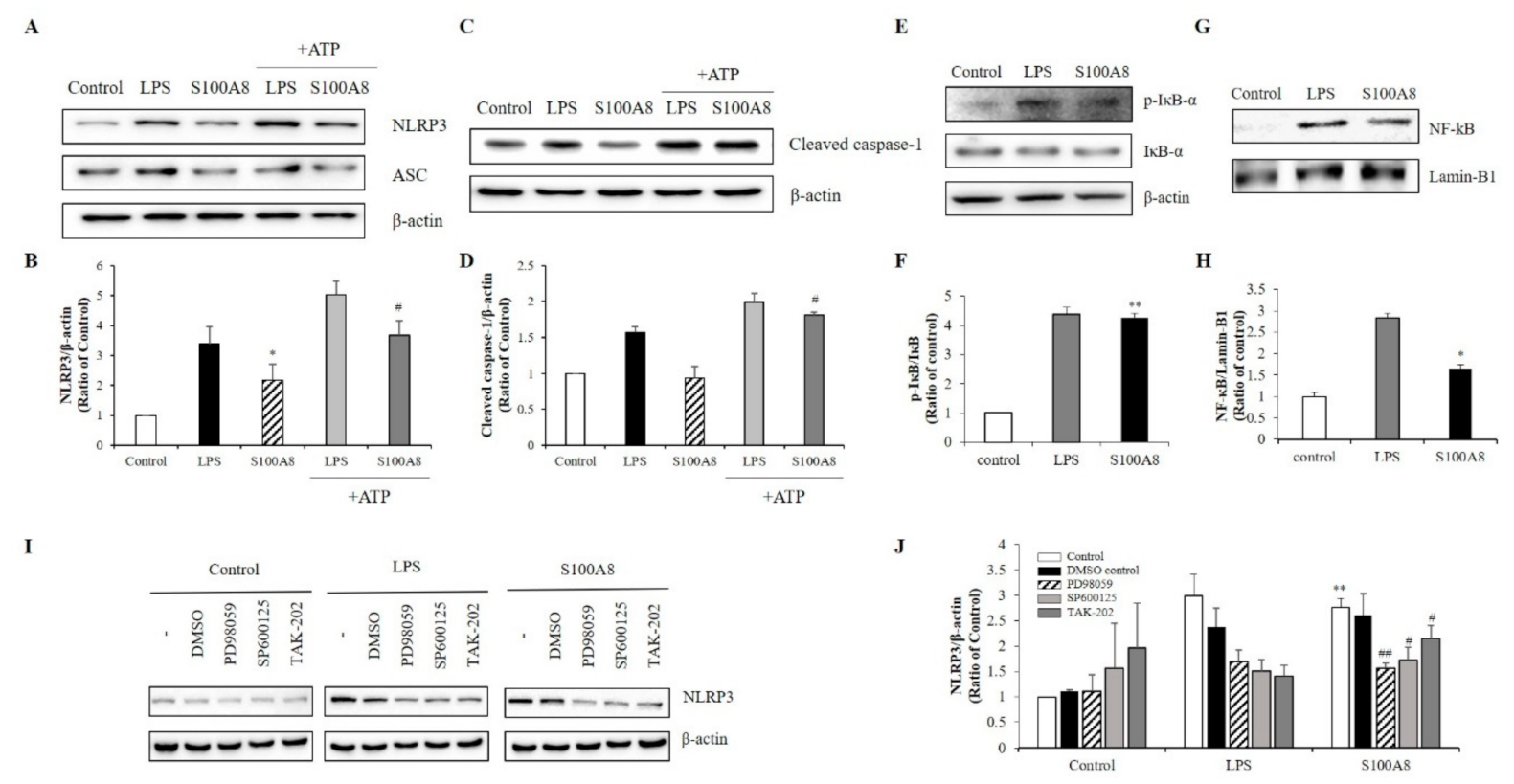
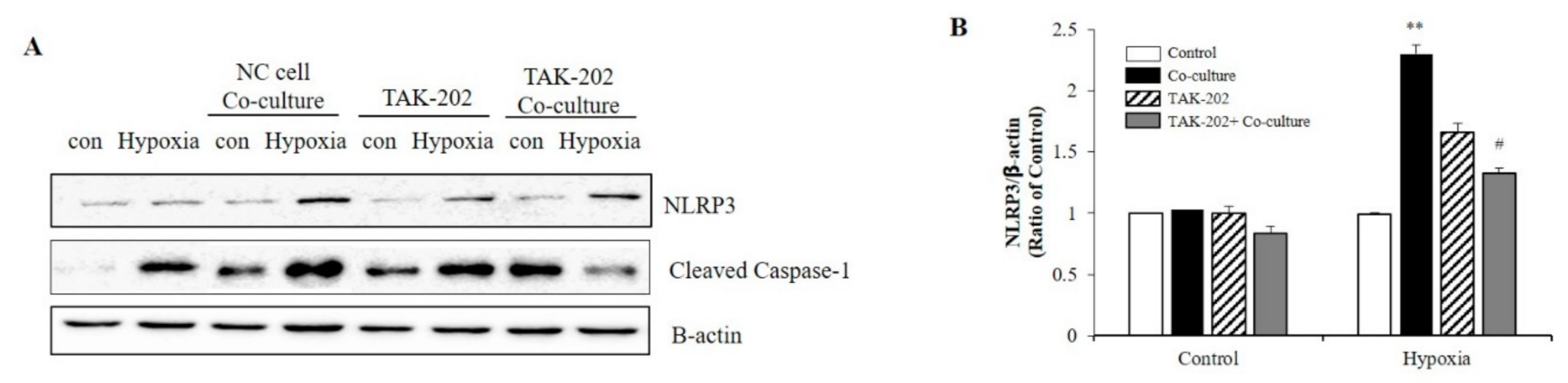
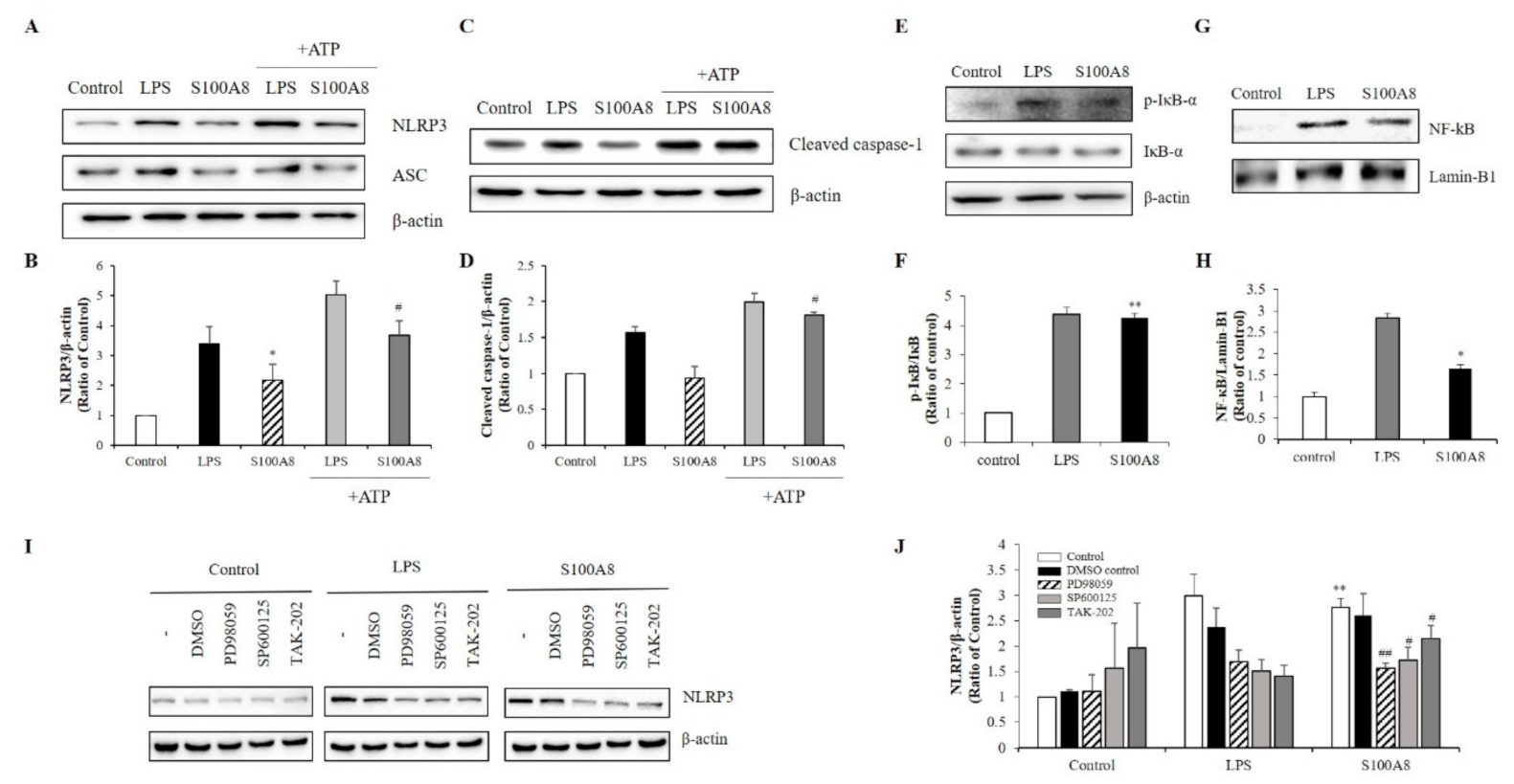
2.3. S100A8 Regulated NLRP3 Inflammasome Priming through TLR4/NF-κB Signaling in BV-2 Cells
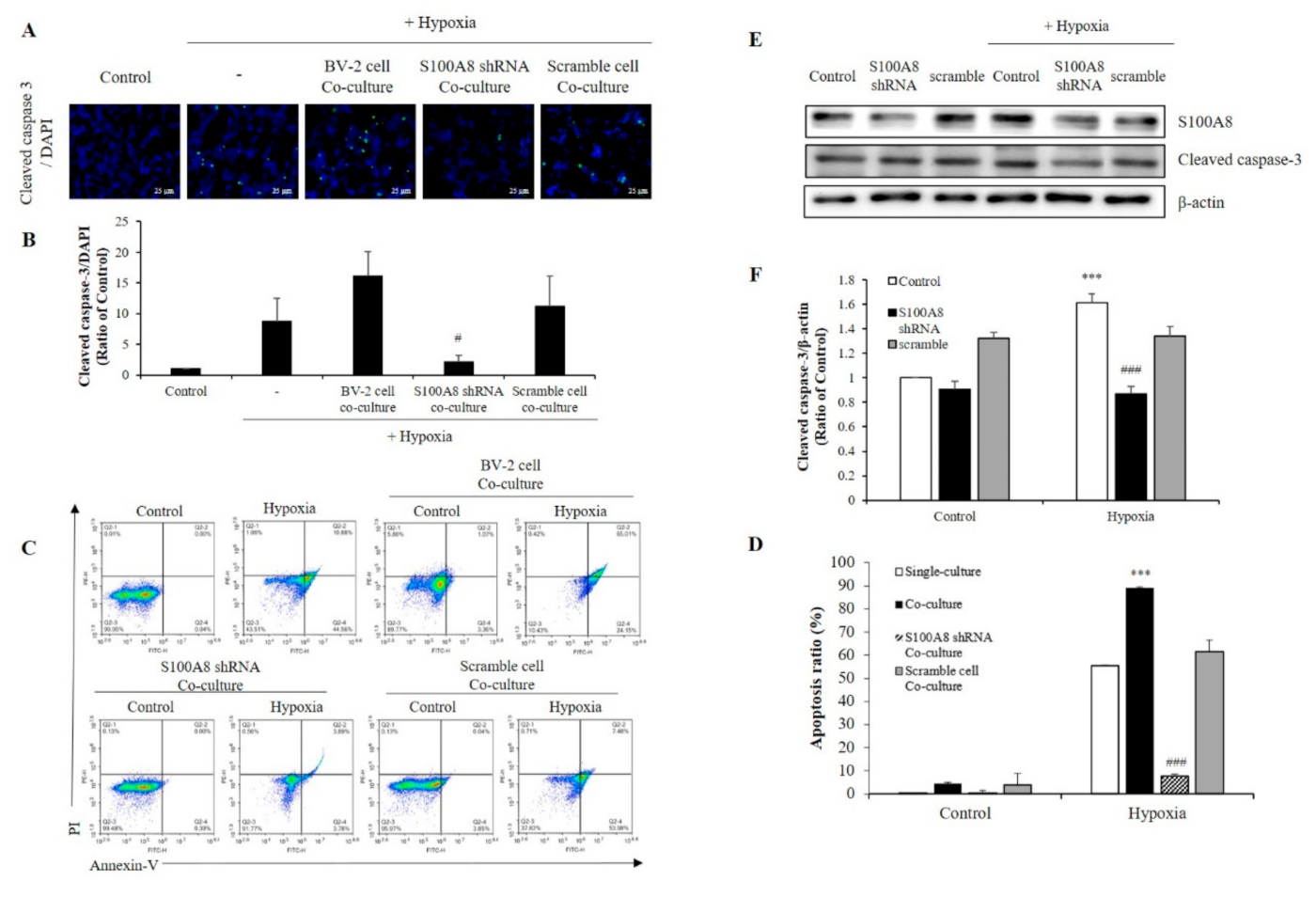
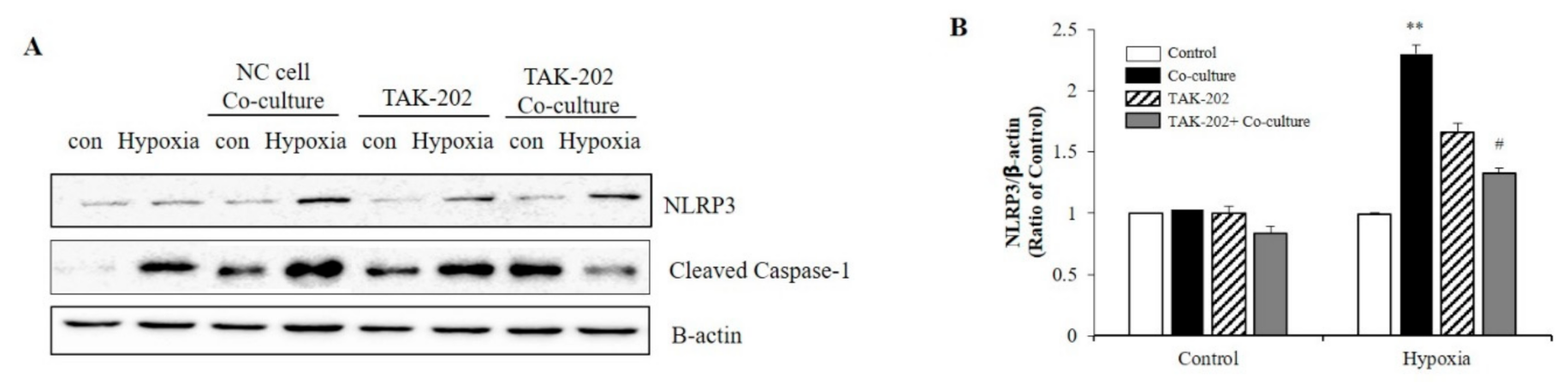
2.4. S100A8 Knockdown on Microglia Attenuated Neuronal Apoptosis by Hypoxia
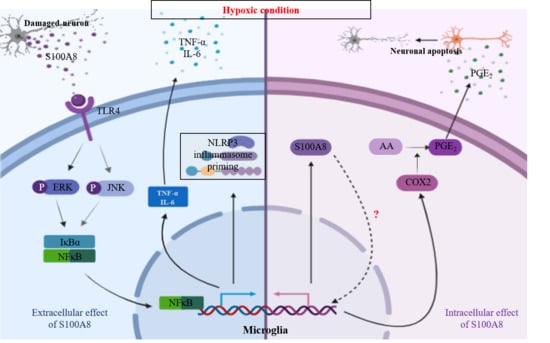
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Plasmids and Transfection
4.4. Measurement of Apoptosis by Flow Cytometry
4.5. Western Blot Analysis
4.6. ELISA
4.7. Immunofluorescence Analysis
4.8. Real-Time Quantitative PCR Analysis
4.9. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DAMP | Damage-associated molecular pattern |
| ERK | Extracellular-signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| HMGB1 | High-mobility group box 1 |
| S100A8 | S100 calcium-binding protein A8 |
| TLR4 | Toll-like receptor-4 |
| MAPK | Mitogen-activated protein kinase |
| NLR | Nucleotide-binding oligomerization domain-like receptor |
| COX-2 | Cyclooxygenase-2 |
| PGE2 | Prostaglandin E2 |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor |
| RAGE | Receptor for advanced glycation end products |
| NF-κB | Nuclear factor- κB |
| AIM2 | Absent in melanoma 2 |
| ASC | Adapter apoptosis-associated speck-like protein |
| AD | Alzheimer’s disease |
| DMEM | Dulbecco’s modified Eagle’s medium |
| PBS | Phosphate-buffered saline |
| FBS | Fetal bovine serum |
| HBSS | Hank’s Balanced Salt Solution |
| ATP | Adenosine 5′-triphosphate disodium salt hydrate |
| SDS | Sodium dodecyl sulfate |
References
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. Bull. World Heal. Organ. 2016, 94, 634. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Park, J.-W.; Hwang, J.-S.; Kim, S.-M.; Lyoo, I.K.; Lee, C.-J.; Han, I.-O. Hypoxia-Induced Neuroinflammation and Learning–Memory Impairments in Adult Zebrafish Are Suppressed by Glucosamine. Mol. Neurobiol. 2018, 55, 8738–8753. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bok, S.; Kim, Y.E.; Woo, Y.; Kim, S.; Kang, S.J.; Lee, Y.; Park, S.K.; Weissman, I.L.; Ahn, G.O. Hypoxia-inducible factor-1α regulates microglial functions affecting neuronal survival in the acute phase of ischemic stroke in mice. Oncotarget 2017, 8, 111508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef]
- Tsuji, S.; Di Martino, E.; Mukai, T.; Tsuji, S.; Murakami, T.; Harris, R.A.; Blomgren, K.; Åden, U. Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J. Neuroinflamm. 2020, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Guadagno, J.; Xu, X.; Karajgikar, M.; Brown, A.; Cregan, S. Microglia-derived TNF α induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member Puma. Cell Death Dis. 2013, 4, e538. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, L.-Y.; Lian-Yan, D.; Hong, Z.; Wei, W.-S. The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor. J. Neuroinflamm. 2012, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Guo, X.; Zhang, J.; Zhou, H.; Sun, B.; Feng, J. DNA binding protein HMGB1 secreted by activated microglia promotes the apoptosis of hippocampal neurons in diabetes complicated with OSA. Brain Behav. Immun. 2018, 73, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Shichita, T.; Ito, M.; Yoshimura, A. Post-ischemic inflammation regulates neural damage and protection. Front. Cell. Neurosci. 2014, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Concepcion, K.; Meng, X.; Zhang, L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog. Neurobiol. 2017, 159, 50–68. [Google Scholar] [CrossRef]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, L.; Wang, Y.; Zhou, N.; Zhen, F.; Zhang, Y.; Qu, X.; Fan, H.; Liu, S.; Chen, Y.; et al. S100A8/A9 induces microglia activation and promotes the apoptosis of oligodendrocyte precursor cells by activating the NF-κB signaling pathway. Brain Res. Bull. 2018, 143, 234–245. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Fann, D.Y.-W.; Lim, Y.-A.; Cheng, Y.-L.; Lok, K.-Z.; Chunduri, P.; Baik, S.-H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.-G.; et al. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.-C.; Cesaro, A.; Chapeton-Montes, J.; Tardif, M.; Antoine, F.; Girard, D.; Tessier, P.A. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-κB 1. PLoS ONE 2013, 8, e72138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.S.; Mann, M.; Dubois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonazzi, A.; Mastyugin, V.; Mieyal, P.A.; Dunn, M.W.; Laniado-Schwartzman, M. Regulation of Cyclooxygenase-2 by Hypoxia and Peroxisome Proliferators in the Corneal Epithelium. J. Biol. Chem. 2000, 275, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aïd, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in Inflammatory and Degenerative Brain Diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Hu, Q.; Wang, H.; Yang, H.; Gao, F.; Ren, H.; Chen, D.; Fu, C.; Ying, Z.; Zhen, X.; et al. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis. 2015, 6, e1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Pernaute, R.; Ferree, A.; Cooper, O.; Yu, M.; Brownell, A.-L.; Isacson, O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. J. Neuroinflamm. 2004, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lodeiro, M.; Puerta, E.; Ismail, M.-A.-M.; Rodriguez-Rodriguez, P.; Rönnbäck, A.; Codita, A.; Parrado-Fernandez, C.; Maioli, S.; Gil-Bea, F.; Merino-Serrais, P.; et al. Aggregation of the inflammatory S100A8 precedes Aβ plaque formation in transgenic APP mice: Positive feedback for S100A8 and Aβ productions. J. Gerontol. Ser. A 2017, 72, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Vijitruth, R.; Liu, M.; Choi, D.-Y.; Nguyen, X.V.; Hunter, R.L.; Bing, G. Cyclooxygenase-2 mediates microglial activation and secondary dopaminergic cell death in the mouse MPTP model of Parkinson’s disease. J. Neuroinflamm. 2006, 3, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Vissel, B. Amyloid β: One of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate A lzheimer’s disease. Br. J. Pharmacol. 2015, 172, 3714–3727. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M.; et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, M.; Nakamachi, T.; Rakwal, R.; Shibato, J.; Nakamura, K.; Wada, Y.; Tsuchikawa, D.; Yoshikawa, A.; Tamaki, K.; Shioda, S. Unraveling the ischemic brain transcriptome in a permanent middle cerebral artery occlusion mouse model by DNA microarray analysis. Dis. Models Mech. 2012, 5, 270–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional microglia–neuron communication in health and disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.; Lee, R.V.; Noor, S.; Lee, C.; Le, T.; Iorga, M.; Phillips, J.L.; Murphy, S.; Möller, T.; Weinstein, J.R. Ischemia/reperfusion induces interferon-stimulated gene expression in microglia. J. Neurosci. 2017, 37, 8292–8308. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- Hanisch, U.-K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.S.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Shin, S.; Kim, H.; Han, S.; Kim, K.; Kwon, J.; Kwak, J.-H.; Lee, C.-K.; Ha, N.-J.; Yim, D. Anti-inflammatory function of arctiin by inhibiting COX-2 expression via NF-κB pathways. J. Inflamm. 2011, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655. [Google Scholar] [PubMed]
- Xing, Y.; Wang, R.; Chen, D.; Mao, J.; Shi, R.; Wu, Z.; Kang, J.; Tian, W.; Zhang, C. COX2 is involved in hypoxia-induced TNF-α expression in osteoblast. Sci. Rep. 2015, 5, 10020. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Choi, E.; Lee, J.-S.; Lee, N.R.; Baek, S.Y.; Gu, A.; Kim, D.H.; Kim, I.S. House Dust Mite Allergen Regulates Constitutive Apoptosis of Normal and Asthmatic Neutrophils via Toll-Like Receptor 4. PLoS ONE 2015, 10, e0125983. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, J.S.; Choi, H.-R.; Kim, I.S.; Kim, E.-A.; Cho, S.-W.; Yang, S.-J. Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis. Int. J. Mol. Sci. 2021, 22, 1205. https://doi.org/10.3390/ijms22031205
Ha JS, Choi H-R, Kim IS, Kim E-A, Cho S-W, Yang S-J. Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis. International Journal of Molecular Sciences. 2021; 22(3):1205. https://doi.org/10.3390/ijms22031205
Chicago/Turabian StyleHa, Ji Sun, Hye-Rim Choi, In Sik Kim, Eun-A Kim, Sung-Woo Cho, and Seung-Ju Yang. 2021. "Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis" International Journal of Molecular Sciences 22, no. 3: 1205. https://doi.org/10.3390/ijms22031205