Dynamical Effects in the Optical Response of Carbon Chains

1
Inst.f. Theor. Physik, Univ. Erlangen, Staudtstr. 7, D-91058 Erlangen, Germany
2
Lab. Phys. Quantique, Univiversité Paul Sabatier, 118 Rte de Narbonne, F-31062 Toulouse Cedex, France
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2002, 3(2), 69-75; https://doi.org/10.3390/i3020069
Submission received: 3 October 2001
/
Accepted: 30 January 2002
/
Published: 28 February 2002
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:We discuss the optical response of small carbon chains from the linear to the non linear domain in the framework of Time Dependent Local Density Approximation. We show that even for moderate ionizations, corresponding to a moderately intense excitation, the optical response exhibits significant alteration with respect to the truly linear domain response. This reflects non trivial dynamical effects at the level of electrons.
1. Introduction
The physics of clusters in intense fields, in particular intense lasers fields, is currently motivating many experimental and theoretical studies [1,2,3,4,5,6,7,8,9,10]. Several experiments have been performed on rare gas clusters [3,5,6] as well as on metal clusters [2,4]. In both, rare gas and noble metal clusters, intense fields can easily ionize semi-core electrons which immediately take part into the response of the system. The details of the behavior of these stripped semi-core electrons is not yet fully understood. In particular, it is expected that these electrons remain for a sizable amount of time inside the clusters, and thus actively participate into the further excitation of the system. They can thus be heated up to keV energies and behave as a plasma which can undergo giant dipole oscillations, both effects leading to a complete and very energetic destruction of the system [2,6,11]. The theoretical description of such highly non-linear situations is still at its beginning [6,8,9,10,11]. In particular, a proper understanding of the behavior of the (originally) rather deeply bound electrons requires the use of sophisticated microscopic methods. A good starting point is the fully fledged Time Dependent Local Density Approximation (TDLDA) as developed at various levels of refinements since a few years [8,9,10,11,12,13,14,15] on the basis of Time Dependent Density Functional Theory [16]. A proper treatment of rare gas or noble metal clusters in a non-linear environment involving deeply lying electrons remains on the very edge of these theories. There is such a need to find simpler systems in which core or semi-core electrons might be easily excited. This means to leave the safe grounds of alkaline metals and to consider systems in which binding is no more metallic and involve a sizable fraction of electrons. A typical example is carbon which is of course the focus of numerous studies, as for example in the (linear domain) optical response of small carbon chains [17,18,19,20,21] or the (semi-linear domain) photoelectrons spectra obtained in C60 [22]. For the exploratory investigations we have in mind, we restrict ourselves to the simpler case of small linear carbon chains. Our aim in this paper is thus to study the response of a small carbon chain to a non-linear perturbation with help of a TDLDA approach. Response in the linear domain was studied in detail within the same theory in [21]. Here we aim at considering the onset of non-linear effects due to electronic dynamical effects.
The technicalities of the model we use have been presented in detail elsewhere and we refer the reader to the recent review [10] for thorough discussions. Let us only briefly remind here the basic inputs for completeness. Electrons are treated in TDLDA with the exchange correlation functional of [23]. We use absorbing boundary conditions to remove disturbances from continuum electrons [10]. The interaction between ions and electrons is described using the non-local pseudo-potential of [24]. Ions are treated as classical particles and can be safely kept fixed during the reasonably short time scales on which we explore the electronic dynamics.
For the exploratory studies here we use a simple one-parameter excitation mechanism: an instantaneous boost of the centre-of-mass of the whole electron cloud. It allows a clean spectral analysis in the linear domain and delivers the change of the response with increasing excitation energy [25]. The test case is axially symmetric throughout. We can thus safely use an axially symmetric description of the electron cloud [26,27,28].
As we aim at describing the optical response and its evolution with intensity of the excitation, we focus on electronic observables. The basic observable is here the electronic dipole moment D(t) (with respect to ionic background), which we follow in time. Fourier transforming D(t) from the time to the frequency domain D(ω) allows to define the strength function S(ω) ~ Im(D(ω)) which is directly related to the photoabsorbption cross section and thus provides a direct access to the optical response. Accessing the non-linear domain leads to electron emission and the optical response has to be studied in relation to the degree of ionization which we measure by the number of emitted electrons Nesc which is computed as the number of electrons leaving the computational box, which has been chosen large enough to avoid artifacts from boundaries [25].
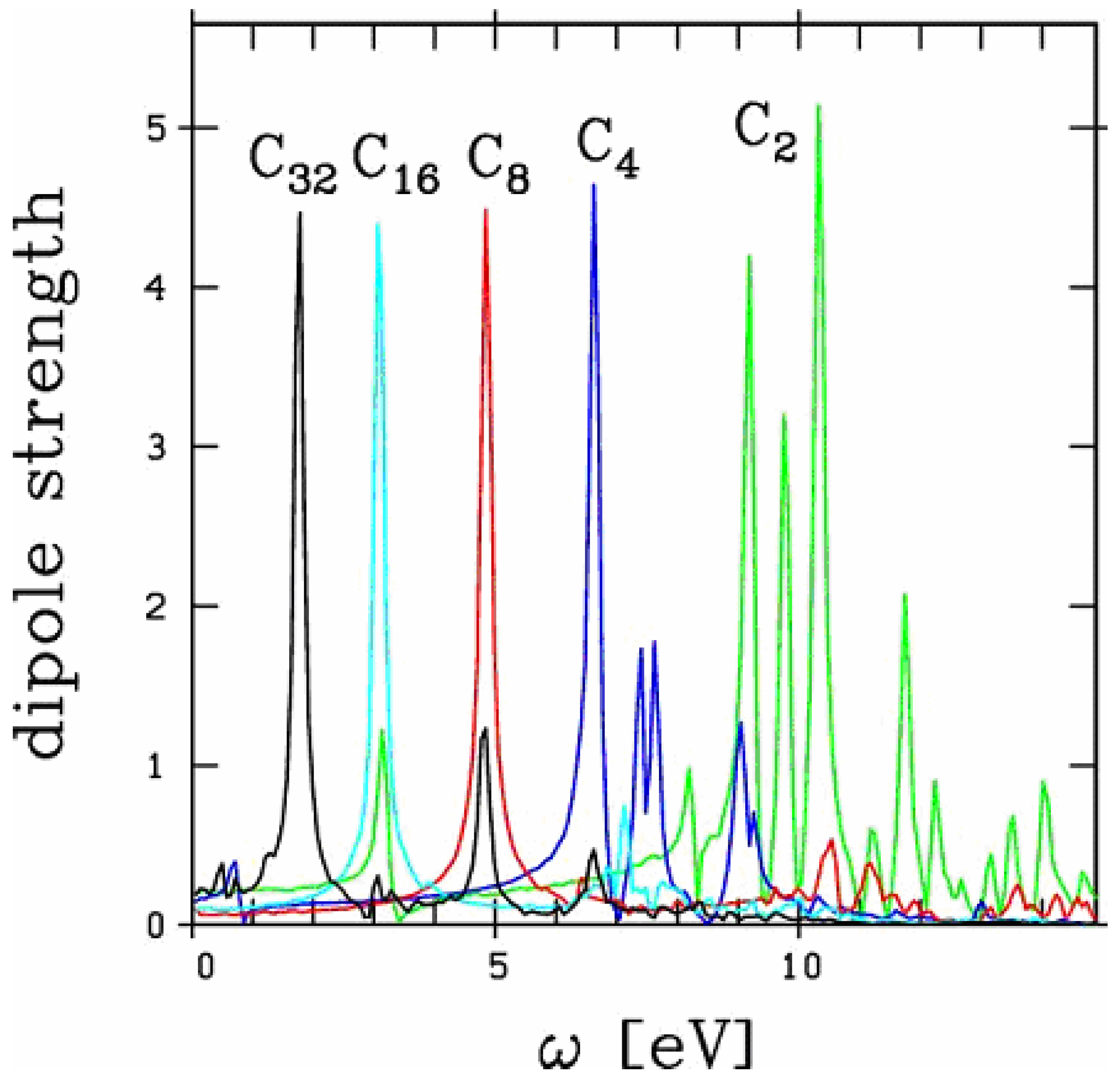
Before directly attacking the case of non-linear excitation we want to check the capability of the method to describe linear optical response in small carbon chains. A typical optical response spectrum is shown is Figure 1. We have collected here the optical response of several carbon chains. Chains
Figure 1.
Longitudinal optical response of linear chains of carbon Cn for n between 2 and 32. The response for energies beyond 15 eV does not exhibit significant peak structures, whatever the system.
Figure 1.
Longitudinal optical response of linear chains of carbon Cn for n between 2 and 32. The response for energies beyond 15 eV does not exhibit significant peak structures, whatever the system.
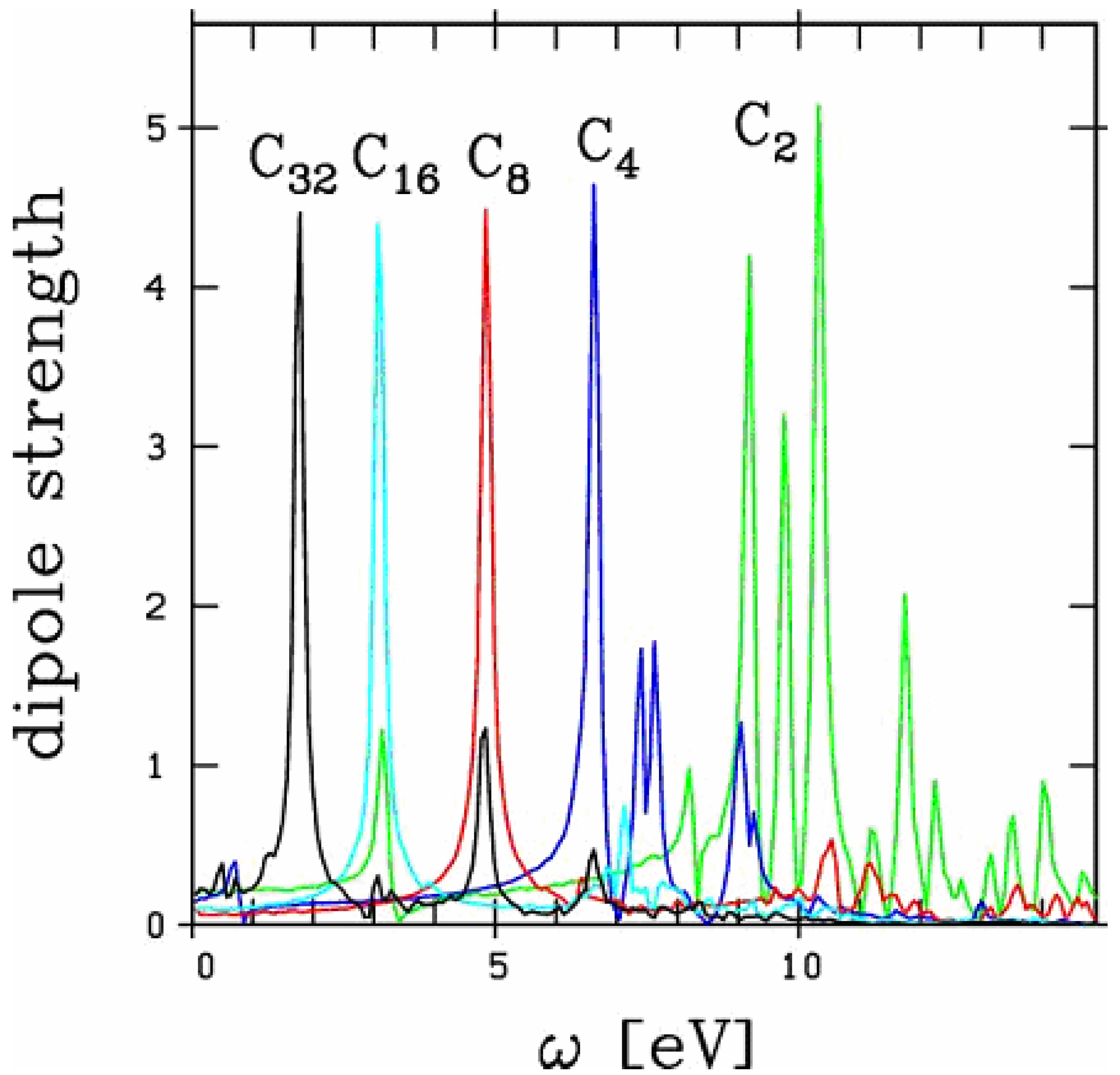
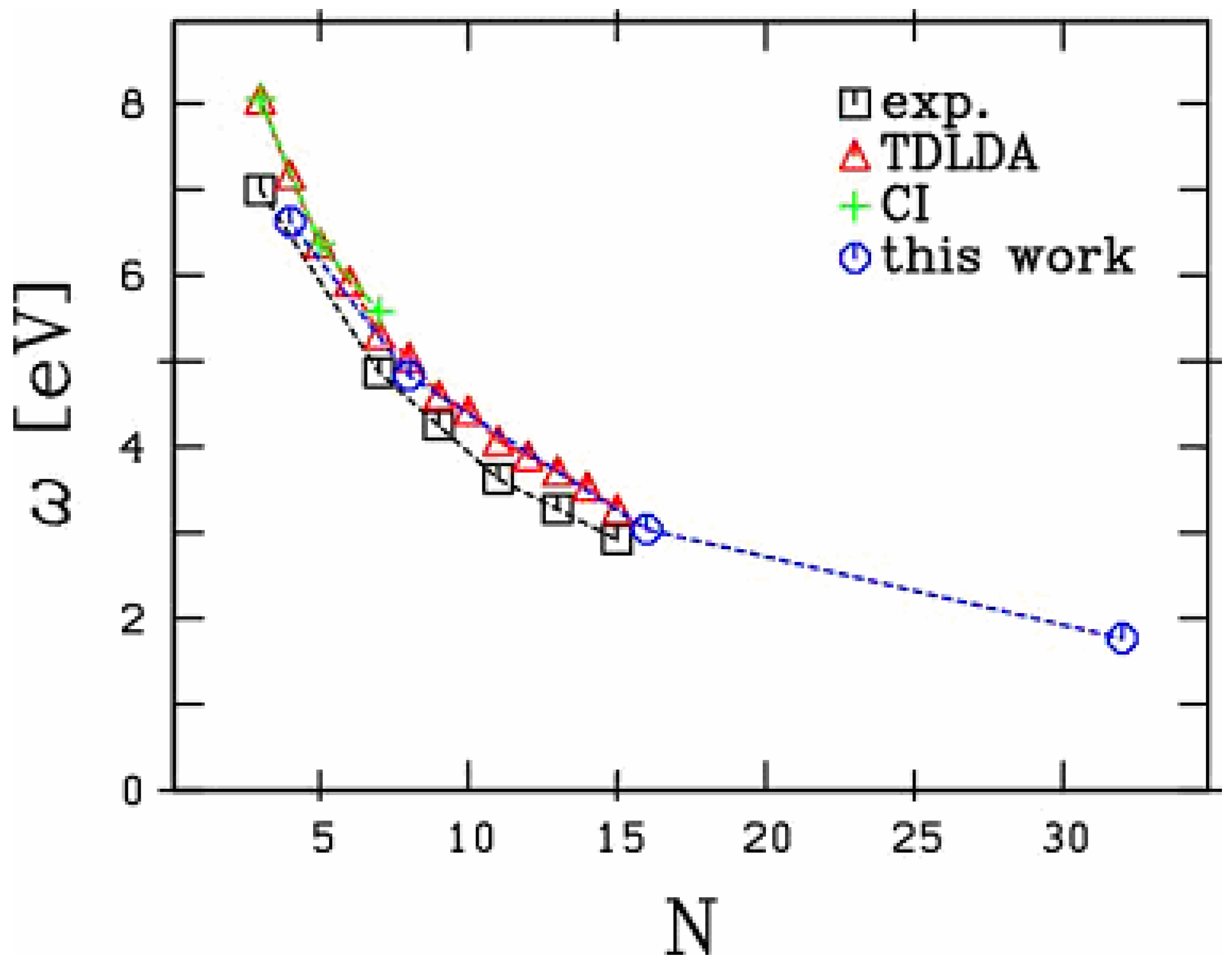
have been unambiguously observed up to 15 C atoms [19,20]. The extension to 32 atoms, as presented in the figure, is thus somewhat academic. Still, the existence of such a structure cannot be fully excluded. We focus here on the longitudinal response, namely the electronic response along the chain. The latter exhibits a strong size dependence as discussed in [21]. In these carbon chains, this response is basically composed of well marked low energy, strongly size dependent, peaks and of a set of very diffuse structures in the 20 eV range. Most of the collective part of the response is concentrated in the low energy structures and we restrict the analysis on this energy range in the following, as in [21]. As can be seen from Figure 1, except for the 2 smallest species (chains with 2 and 4 atoms) the optical response is dominated by one single peak. In small chains, although the response is slightly more fragmented, a well defined peak remains clearly visible. The energy of this peak strongly decreases with size reflecting a clear geometric effect: the longer the chain and thus the participating electron cloud, the longer the oscillation period and hence the smaller the frequency. In order to visualize this effect in a clearest manner we plot in Figure 2 the energy of this main peak as a function of chain size. For completeness we have also reported the experimental results of [19,20] and the theoretical results of [21] (TDLDA calculations) and [17,18] (quantum chemistry CI calculations). The agreement between all calculations and experiments is impressive, in all cases for which the comparison is possible.
Figure 2.
Systematics of the longitudinal mode in small carbon chains. Comparison is performed between experiments [19] and [20], and various calculations: TDLDA from [21], quantum chemistry (CI) calculations from [17] and [18].
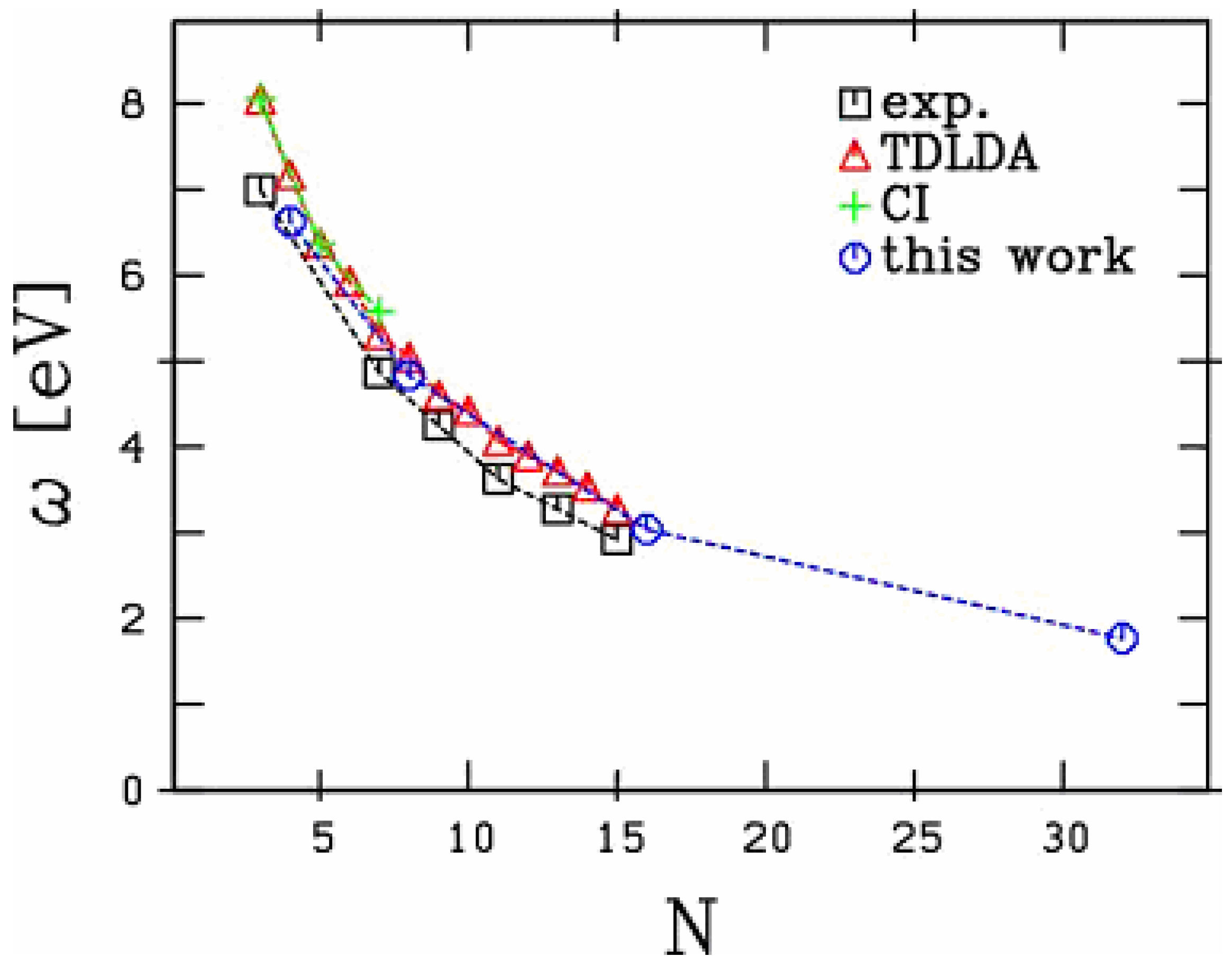
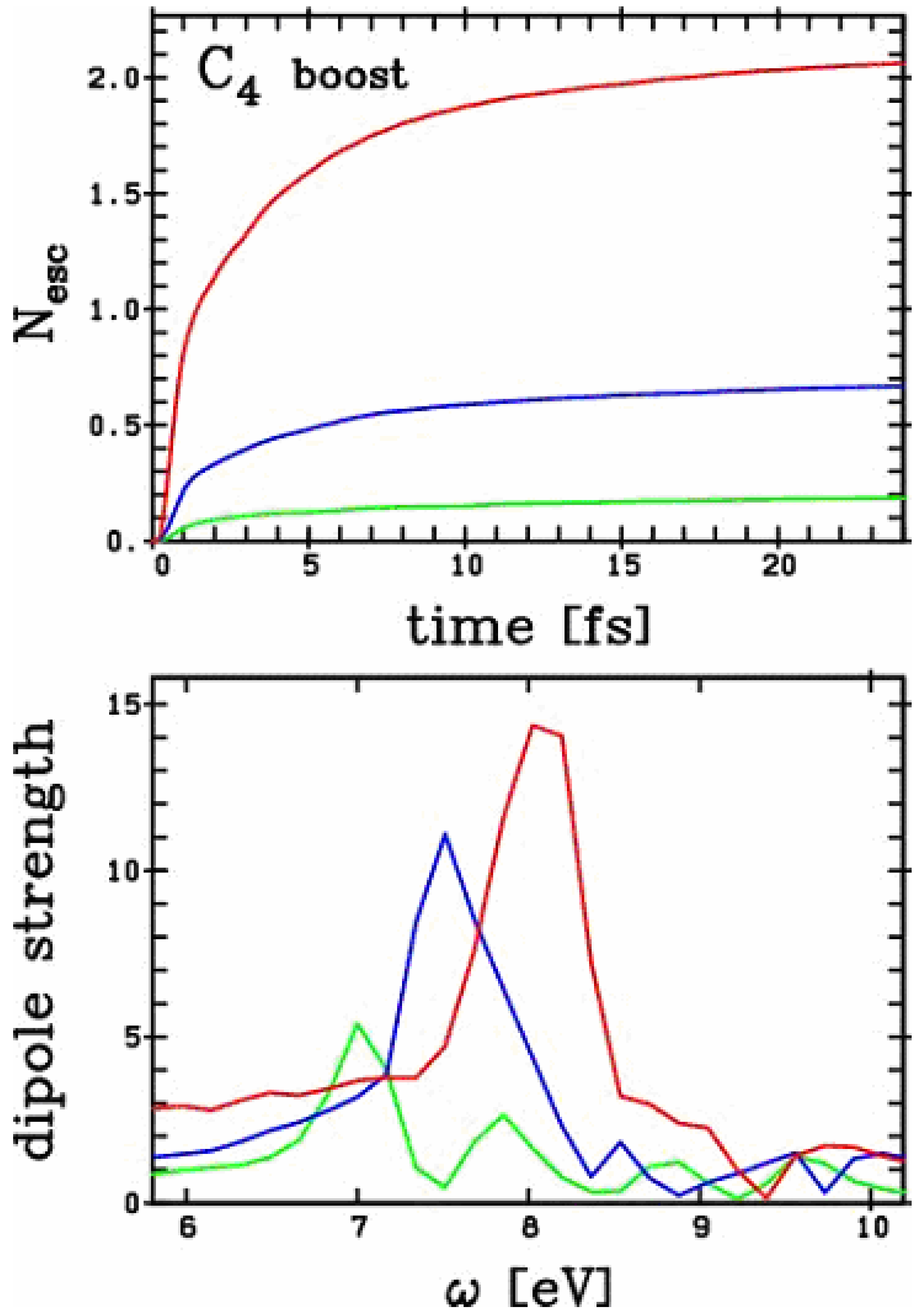
Figure 3.
Optical response (lower panel) and ionisation (upper panel) of a C4 chain to excitations of various strengths of excitation. The initial boost placed the following amounts of energy into the cluster: green = 0.3 Ry, blue = 1.3 Ry, and red = 5.1 Ry.
Figure 3.
Optical response (lower panel) and ionisation (upper panel) of a C4 chain to excitations of various strengths of excitation. The initial boost placed the following amounts of energy into the cluster: green = 0.3 Ry, blue = 1.3 Ry, and red = 5.1 Ry.

We now turn towards an exploration of non-linear excitations. In Figure 3 we have plotted the optical response of a C4 chain for various boosts with increasing strength (doubling the boost with each higher excitation). The increasing violence of the process can be seen from the upper panel of Figure 3 where we plotted the ionization as a function of time for the various cases. We explore a sizable range of non-linearity. Mind that in this chain 16 electrons do participate in the binding, and thus constitute the basic "electron reservoir" on which to build electronic excitations. In the most violent case, as much as two electrons have been stripped from the system. The optical response is significantly altered by this ionization process. We observe a marked blue shift of the major peak with increasing ionization. For example, the peak of the doubly charged chain is more than 1 eV higher in energy than the peak of the neutral chain. We find here an effect similar to the one observed in metal clusters [10], although its amplitude is more marked in the carbon case. However, it is not clear from this analysis how non-trivial the effect is here. The blue shift of the optical response is primarily a simple electrostatic effect reflecting the fact that the more charged the cluster, the stronger the restoring force exerted by the ionic background on the electron cloud. In the case of metal clusters such an electrostatic effect fully explains the blue shift of the peak with increasing ionization, the remaining possible effect due to internal excitation of electron cloud remaining of negligible impact [10].
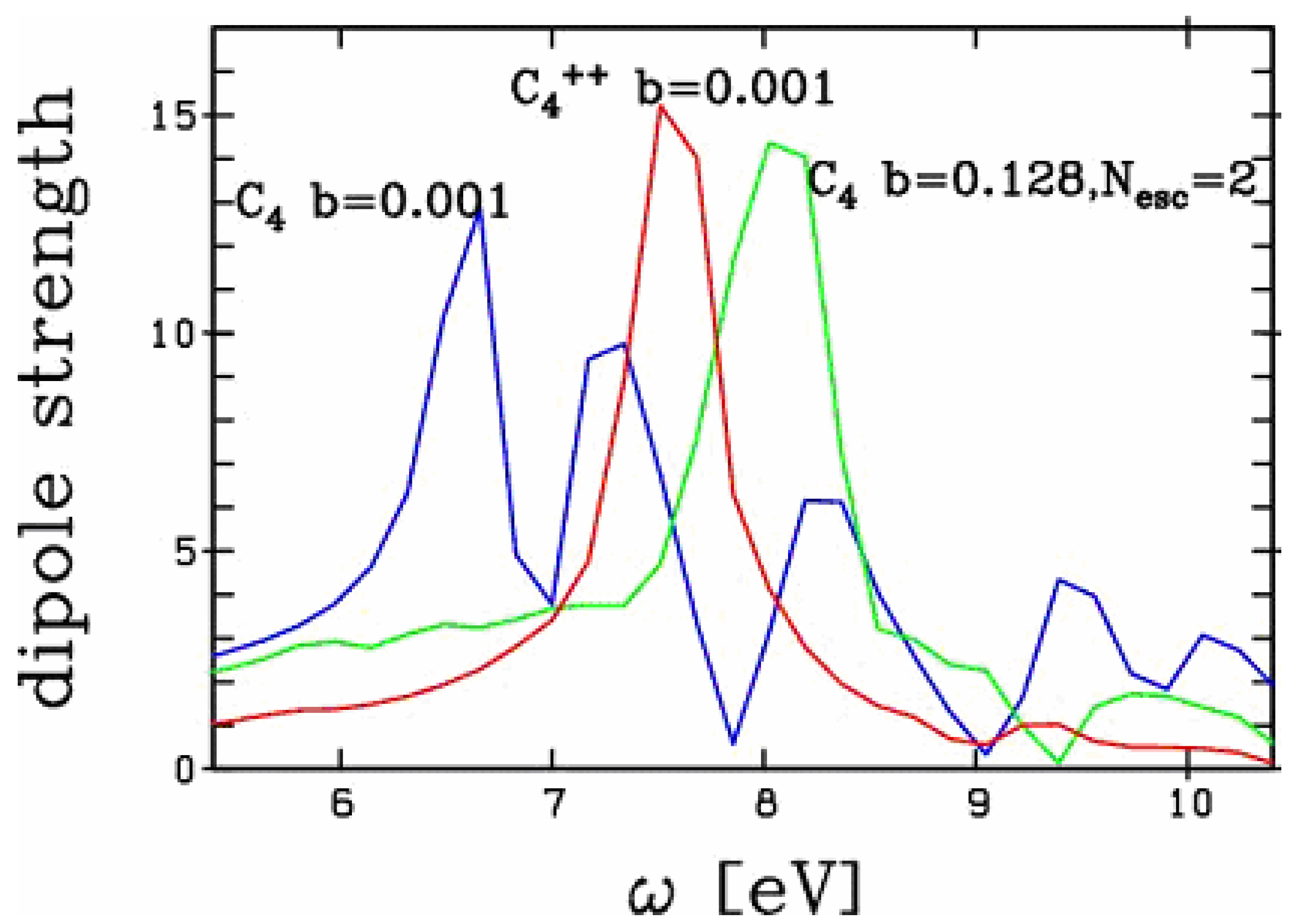
In order to explore whether the blue shift observed in Figure 3 is a pure electrostatic effect or not, we have compared the optical response as obtained by a non-linear excitation of C4 leading to highly excited C4++, to the linear response of ground state C4++. The comparison is presented in Figure 4. For completeness we have also plotted the optical response of ground state C4 in the linear domain. We observe a marked difference between the optical responses of the two doubly ionized clusters: the two peaks differ by 0.5 eV. The charge effect thus only explains about half of the total blue shift of the
Figure 4.
Comparison of the optical responses of C4 to a non-linear excitation (boost b=0.128) leading to an emission of 2 electrons with the response of C4++ to linear domain excitation (boost b= 0.001, no emission). The response of C4 to a linear excitation is also given for completeness.
Figure 4.
Comparison of the optical responses of C4 to a non-linear excitation (boost b=0.128) leading to an emission of 2 electrons with the response of C4++ to linear domain excitation (boost b= 0.001, no emission). The response of C4 to a linear excitation is also given for completeness.
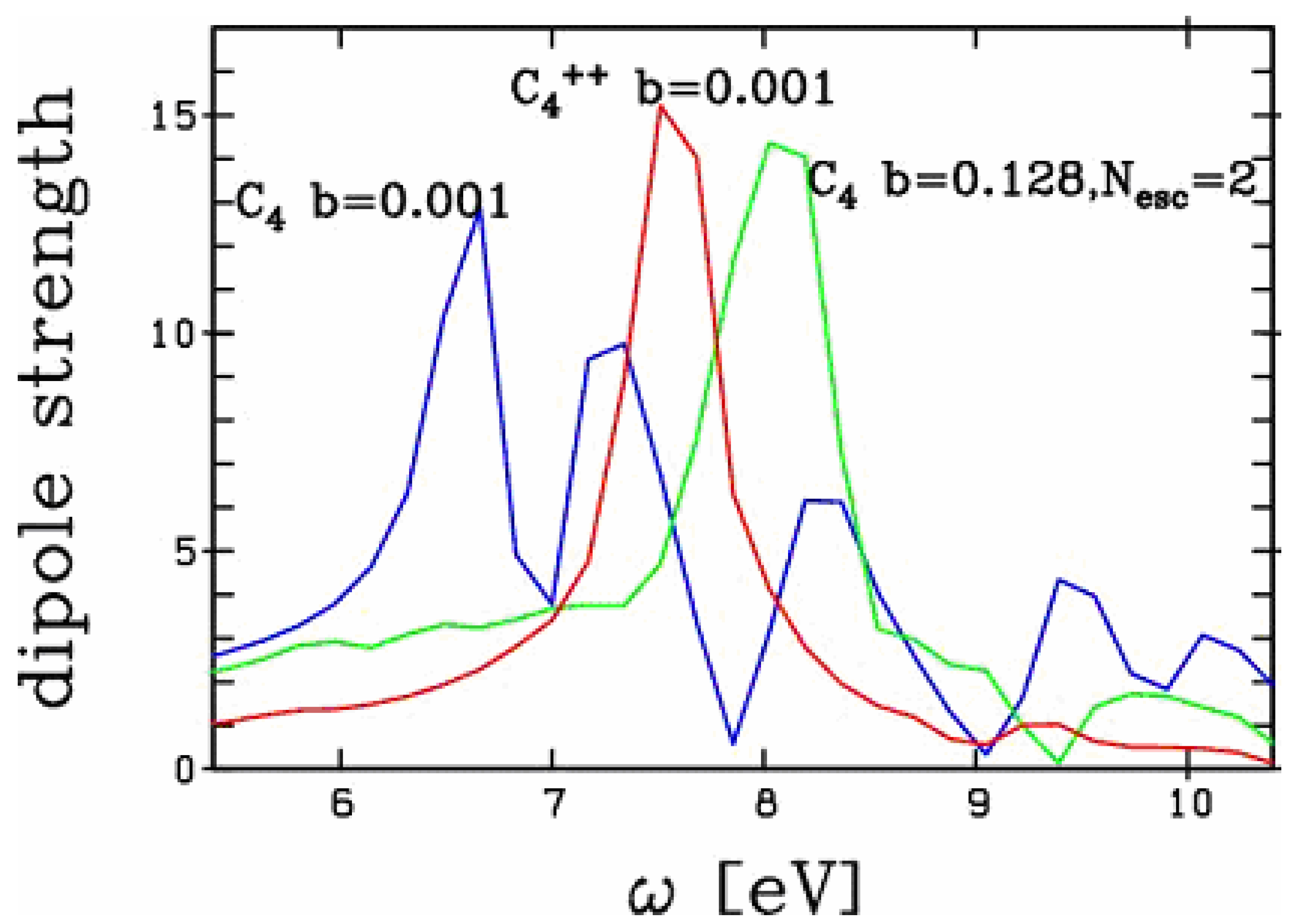
response. Such a behavior is markedly different from the one observed in metal clusters [10]. There is also a marked difference in the single particle energies of the valence electrons in metal clusters as compared to carbon chains. The levels are close together in simple metal clusters while they group into different strengths of binding in carbon chains. For example in the C4 studied here, the p electrons gather close to the Fermi energy ranging actually between -0.5 and -0.7 Ry binding while the s electrons are well separated in energy ranging from -1 to -2 Ry binding energy. The above results suggest that strong dynamical effects take place in the excited electronic cloud, following a non-linear excitation. This might reflect dynamical activation of originally deeper bound electrons. A more thorough analysis is here necessary in order to draw more definite conclusions.
In this paper we have analyzed the optical response of small carbon chains, scanning linear as well as non-linear excitations. The carbon chains offer simple model cases for analyzing the response of well bound systems with "valence shell" electrons at very different degrees of binding. Such systems, beyond their intrinsic interest, can also help understanding the behavior of rare gas and noble metal clusters subject to intense laser fields, in which the behavior of semi-core electrons is not fully understood. We have found that in the non-linear domain, the "valence" electrons react in a particularly complex way, the response involving both sizable electron emission and strong dynamical effects of the remaining electrons. A complete analysis of the situation, in order to identify the origin of such effects goes beyond the scope of this paper. Work in this direction is in progress.
Acknowledgements
The authors thank the French-German exchange program PROCOPE, contract number 99074, and Institut Universitaire de France for financial support during the realization of this work.
References
- Ditmire, T.; Tisch, J.W.G.; Springate, E.; Mason, M.B.; Hay, N.; Smith, R.A.; Marangos, J.; Hutchinson, M.H.R. Nature 1997, 386, 54.
- Köller, L.; Schumacher, M.; Köhn, J.; Teuber, S.; Tiggesbäumker, J.; Meiwes-Broer, K. H. Phys. Rev. Lett. 1999, 82, 3783. [CrossRef]
- Lezius, M.; Dobosz, S.; Normand, D.; Schmidt, M. Phys. Rev. Lett. 1998, 80, 261. [CrossRef]
- Schlipper, R.; Kusche, R.; Issendorff, B.; Haberland, H. Phys.Rev.Lett. 1998, 80, 1194. [CrossRef]
- Springate, E.; Hay, N.; Tisch, J. W. G.; Mason, M. B.; Ditmire, T.; Hutchinson, M. H. R.; Marangos, J. P. Phys. Rev. A 2000, 61, 063201.
- Mcpherson, A.; Thompson, B.D.; Borisov, A.B.; Boyer, K.; Rhodes, C.K. Nature 1994, 370, 631.
- Rose–Petruck, C.; Schafer, K. J.; Wilson, K. R.; Barty, C. P. J. Phys. Rev. A 1997, 55, 1182.
- Daligault, J.; Guet, C. Phys. Rev. A 2001, 64, 043203. [CrossRef]
- Ullrich, C. A.; Reinhard, P.-G.; Suraud, E. J. Phys. B 1998, 31, 1871. [CrossRef]
- Calvayrac, F.; Reinhard, P.-G.; Suraud, E.; Ullrich, C. Phys.Rep. 2000, 337, 493. [CrossRef]
- Suraud, E.; Reinhard, P. G. Phys. Rev. Lett. 2000, 85, 2296. [CrossRef] [PubMed]
- Yabana, K.; Bertsch, G. F. Phys. Rev. B 1996, 54, 4484. [CrossRef]
- Gross, M.; Guet, C. Z. Phys D 1995, 33, 289.
- Feret, L.; Suraud, E.; Calvayrac, F.; Reinhard, P.-G. J. Phys B 1996, 29, 4477.
- Calvayrac, F.; Reinhard, P.-G.; Suraud, E. Phys. Rev. B 1995, 52, R17056. [CrossRef]
- Density Functional Theory, NATO ASI Series B; Gross, E.K.U.; Dreizler, R.M. (Eds.) Plenum Press: New York, 1995; Vol. 337.
- Pacchioni, G.; Koutecky, J. J. Chem. Phys. 1988, 88, 1066.
- Kolbuszewski, M. J. Chem. Phys. 1995, 102, 3679.
- Chang, K.; Graham, W. J. Chem. Phys. 1982, 77, 4300.
- Forney, D.; Freivogel, P.; Grutter, M.; Maier, J.P. J. Chem. Phys. 1996, 104, 4954.
- Yabana, K.; Bertsch, G. F. Zeit. Phys. D 1997, 42, 219. [CrossRef] [Green Version]
- Campbell, E.; Hansen, K. Phys. Rev. Lett. 2000, 84, 2128. [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Phys. Rev. B 1992, 45, 13244. [CrossRef]
- Goedeker, S.; Teter, M.; Hutter, J. Phys. Rev. B 1996, 54, 1703.
- Calvayrac, F.; Reinhard, P.-G.; Suraud, E. Ann. Phys. (N.Y.) 1997, 255, 125. [CrossRef]
- Montag, B.; Reinhard, P.-G. Phys. Lett. A 1994, 193, 380. [CrossRef]
- Montag, B.; Reinhard, P.-G. Z. Phys. D 1995, 33, 265. [CrossRef]
- Calvayrac, F.; Reinhard, P.-G.; Suraud, E. Eur. Phys. J. D 1999, 9, 389.
© 2002 by MDPI (http://www.mdpi.org), Basel, Switzerland.
Share and Cite
MDPI and ACS Style
Berkus, T.; Reinhard, P.-G.; Suraud, E. Dynamical Effects in the Optical Response of Carbon Chains. Int. J. Mol. Sci. 2002, 3, 69-75. https://doi.org/10.3390/i3020069
AMA Style
Berkus T, Reinhard P-G, Suraud E. Dynamical Effects in the Optical Response of Carbon Chains. International Journal of Molecular Sciences. 2002; 3(2):69-75. https://doi.org/10.3390/i3020069
Chicago/Turabian StyleBerkus, T., P.-G. Reinhard, and E. Suraud. 2002. "Dynamical Effects in the Optical Response of Carbon Chains" International Journal of Molecular Sciences 3, no. 2: 69-75. https://doi.org/10.3390/i3020069