Gas–Phase Studies of Spin–Spin Coupling Constants

Laboratory of NMR Spectroscopy, Department of Chemistry, University of Warsaw, ul. Pasteura 1, 02-093 Warszawa, Poland
Int. J. Mol. Sci. 2003, 4(3), 135-142; https://doi.org/10.3390/i4030135
Submission received: 30 August 2002
/
Accepted: 4 November 2002
/
Published: 25 February 2003
(This article belongs to the Special Issue Nuclear Magnetic Resonance Spin-Spin Coupling Constants - Calculations and Measurements - Part I)
Abstract
:Recent results of experimental spin-spin coupling constants are reviewed and their relation to ab initio calculations is discussed. It is shown that the NMR measurements of spin-spin coupling are density dependent in the gas phase. The extrapolation to the zero-density limit is required in order to obtain the Jo coupling constants which are free from intermolecular interactions. Such coupling constants can be used as the experimental standards for any comparison with the results of appropriate calculations. It is also pointed out that the effects of the rotational and vibrational motion of nuclei in a molecule can be estimated completely only by theoretical methods.
Introduction
The nuclear magnetic shielding and indirect spin-spin coupling constants measured by nuclear magnetic resonance (NMR) spectra are essentially of importance in qualitative and structural analysis of chemical compounds. The theoretical calculations of NMR parameters play a very important role at this point. The analysis has already reached quantitative accuracy for small molecules due to both the modern NMR techniques and precise calculations [1]. At present shielding calculations are relatively easy, and can be carried out routinely at the SCF level providing often fairly accurate results. The calculations of spin-spin coupling demand more sophisticated treatments, but now they are becoming available [2,3] even though their accuracy is not always easy to estimate. It is an urgent question, which experimental data can straightforward be compared with the theoretical results of spin-spin coupling? The answer is not easy, NMR spectra of isotropic species supply only averaged values of coupling known as the spin-spin coupling constants. The NMR measurements performed for molecules dissolved in liquid crystals give additional information on the anisotropy of spin-spin coupling tensors. More sophisticated experiments are also needed to determine the absolute (or at least relative) signs of spin-spin coupling constants. All the latter experimental data still contain the contributions from molecular interactions and intramolecular nuclear motion and these effects must be accounted if a reliable comparison of theoretical and experimental coupling constants is required.
Solvent and rovibrational effects
It is well known that spin-spin coupling constants measured in NMR spectra are solvent-dependent. The dependence can be negligibly small for some chemical compounds but it is always present in every measurement performed for a macroscopic sample. Molecular interactions are responsible for the modification of coupling constants and this effect is certainly smaller for gases than for liquids. Early measurements in the gas phase have been described by Laszlo [4]. More results have been shown in a review of solvent effects on nuclear spin-spin coupling constant provided by Barfield et al. [5]. These data are sometimes used for the verification of ab initio calculations performed for isolated molecules. Measurements in the gas phase certainly give better approximation than any other experiments made for liquids. However, intermolecular interactions and intramolecular nuclear motions are obviously present in the gas phase and they influence experimental results. Fig. 1 gives a qualitative description of this problem. As seen, the intermolecular effects can be eliminated from the experimental coupling constants if NMR measurements are performed in the gas phase and extrapolated to the zero-density limit.
Figure 1.
Modifications of spin-spin coupling constants due to internal rovibrational motion of a molecule (1) → (2) and molecular interactions (2) → (3).
Figure 1.
Modifications of spin-spin coupling constants due to internal rovibrational motion of a molecule (1) → (2) and molecular interactions (2) → (3).
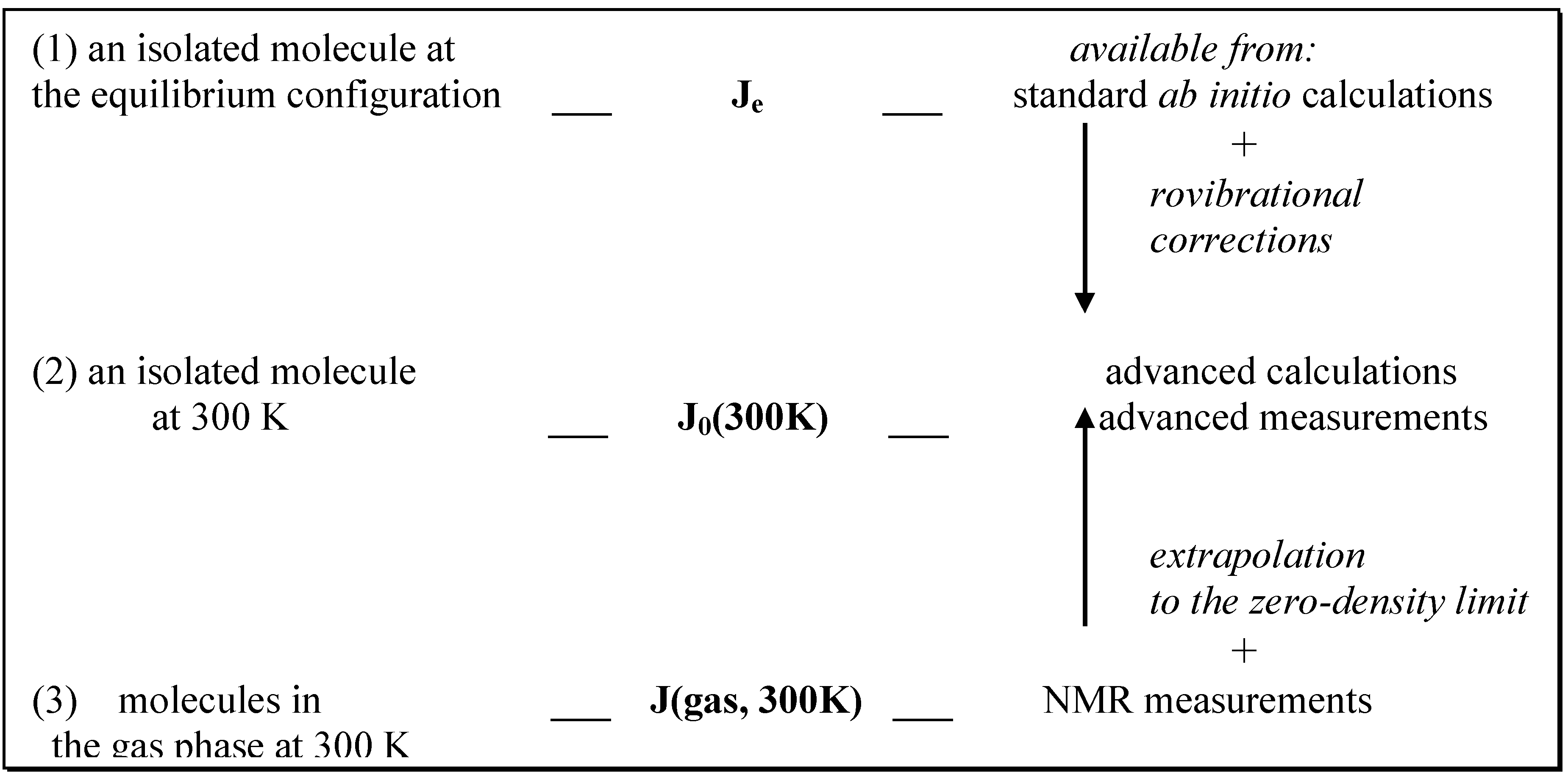
This can be done at room temperature or even better at 300 K. The small increase of temperature is really useful because it allows the stabilization of temperature without any cooling system. The next step belongs to theoretical studies. Spin-spin coupling constants calculated for the equilibrium geometry of a molecule require rovibrational corrections and this has already been done for some molecules, i.e. hydrogen [6], acetylene [7,8], water [9], methane [10] and silane [11]. As an example, Table 1 shows the magnitude of solvent effects and rovibrational corrections for the spin-spin coupling constants of 1,2-13C-acetylene [12]. As shown, these contributions are significant, especially for the one-bond 1J(13C-13C) coupling constants, and cannot be ignored when theoretical and experimental coupling constants are compared.

{kind=link}
Table 1.
Experimental spin-spin coupling constants for 1,2-13C-acetylene in Hz [12].
| J | Je a | J0(300K)b | Jsoln.(300K)c |
|---|---|---|---|
| 1J(13C-13C) | 185.04 | 174.78 | 165.8 |
| 1J(13C-1H) | 242.69 | 247.56 | 248.3 |
| 2J(13C-1H) | 53.84 | 50.14 | 49.9 |
| 3J(1H-1H) | 10.89 | 9.62 | 9.6 |
Density dependence of coupling constants
In a gas of low density, the nuclear spin-spin coupling (J) can be written as an expansion in powers of density (ρ):
where J0(T) is the spin-spin coupling constant for an isolated molecule, T is temperature and J1(T) is a measure of the effects on spin-spin coupling due to binary collisions. The higher order terms, starting from J2(T), are negligible for low-density samples. Then the density dependence is linear and the two spin-spin coupling parameters (J0(T) and J1(T)) are easily available from eq.(1). This approach has first been applied by Jameson and Reger [13] when the one-bond 29Si - 19F coupling was studied for pure SiF4 and its gaseous solutions in CH4, HCl and CO2. The authors obtained the 1Jo(29Si-19F) and 1J1(29Si-19F) values for pure SiF4 and the appropriate 1J1AB(29Si-19F) values for the gaseous solutions (the 1J1AB parameter is defined by eq.(2) later in this text). It seems that the precision of these early measurements was considerably overestimated [13], especially for the 1J1(29Si-19F) and 1J1AB(29Si-19F) parameters but the original results have been cited in Table 2 of this review. Within the experimental error no density dependence was observed for the one-bond coupling constants in CH4, SiH4 and PF3 [13]. Jameson et al. [14] have analyzed the broad 1:3:3:1 multiplet in the 11B NMR spectrum of gaseous BF3. The dependence on density and temperature has been found for the 19F - 11B spin-spin coupling constant. The 1Jo(19F-11B) and 1J1(19F-11B) parameters at 300 K are shown in Table 2. Recently, the large density dependence has been measured for the 19F - 13C spin-spin coupling in pure CD3F [15]. Its second virial coefficient of eq. (1) is equal to -1008(72) (Hz ml mol-1) and it is almost an order of magnitude larger than the previously known values of J1, cf. Table 2. This example is pronounced and clearly shows that the extrapolation to the zero-density limit is absolutely necessary if we need to obtain the Jo coupling constant. It is interesting that the other coupling constants of CD3F (1J(13C-2H) and 2J(19F-2H)) have no distinct dependence on density.
J(ρ,T) = J0(T) + J1(T) + J2(T) + ...
Table 2.
Spin-spin coupling constants at the zero-density limit (nJo) and their dependence on density in pure gaseous compounds (nJ1) and in binary mixtures (nJ1AB).
| Coupling constant | Molecule | Gas solvent | Jo [Hz] | J1 or J1AB [Hz ml mol-1] | Method, temperature | Reference |
|---|---|---|---|---|---|---|
| 1J(29Si-19F) | SiF4 | SiF4 CH4 HCl CO2 | -169.0(1)a | -158(22)a -239(25)a -172(5)a -123(15)a | 19F | [13] |
| 1J(19F-11B) | BF3 | BF3 | 17.77 | 154 | 11B, 300 K | [14] |
| 1J(13C-13C) | 13C2H2 | Xe CO2 | 174.77(2)174.78(2) | -30(20) -301(20) | 1H and 13C, 300 K | [12] |
| 1J(19F-13C) | CD3F | CD3F | 160.72(5) | -1008(72) | 13C, 300 K | [15] |
| 1J(13C-2H) | CD3F | CD3F | 22.45(5) | 43(46) | 13C, 300 K | [15] |
| 2J(19F-2H) | CD3F | CD3F | 7.20(5) | 0 | 19F, 300 K | [15] |
| 1J(15N-13C) | CH313C15N | SF6 CO2 | -16.20(1) -16.23(2) | -62(14) -191(44) | 15N and 13C, 300 K | [16] |
| 2J(13C-1H) | CH313C15N | SF6 CO2 | -10.18(2) -10.19(2) | -13(37) 6(46) | 1H and 13C, 300 K | [16] |
| 3J(15N-1H) | CH313C15N | SF6 CO2 | -1.34(2) -1.43(9) | -325(77) -134(171) | 15N and 1H, 300 K | [16] |
| 1J(13C-1H) | 13CH313CN | SF6 CO2 | 134.03(1) 134.05(1) | 95(24) 148(23) | 1H and 13C, 300 K | [17] |
| 1J(13C-13C) | 13CH313CN | SF6 CO2 | 60.12(5) 60.12(3) | -86(87) -151(56) | 13C, 300 K | [17] |
a Assuming that the one-bond 1J(29Si-19F) coupling constant is negative.
Many results shown in Table 2 have been obtained for gaseous binary mixture where the solute gas (A) was observed at constant pressure and the density dependence was due to the solvent gas (B). Generally, nuclear spin-spin coupling is modified by interactions of pairs of molecules in the binary mixtures of gases (A and B) as follows:
where Jo(XY) is the spin-spin coupling between X and Y nuclei at the zero-density limit and J1AA(XY), J1AB(XY) are solely due to intermolecular effects in the binary collisions of A-A and A-B molecules, respectively. Usually, the density of A is kept sufficiently low for eq. (2) to simplify it to
J(XY) = Jo(XY) + J1AA(XY)ρA + J1AB(XY)ρB + ....
J(XY) = Jo(XY) + J1AB(XY)ρB
This experimental method is especially attractive when the solute can produce a strong NMR spectrum, then the approximation described by eq. (3) can easily be achieved and the small quantity of solute permits the exact measurements of spin-spin coupling constants. Table 2 contains the 1Jo(13C-13C) and 1J1AB(13C-13C) parameters for the one-bond coupling in 1,2-13C-enriched acetylene [12]. As seen, the same coupling constant has different density dependence in the investigated solvents (cf. 1J1AB for Xe and CO2) and the constant for an isolated molecule (1Jo) remains the same confirming the accuracy of the measurement. Similar results have been obtained for the 2 isotopomers of acetonitrile: CH313C15N [16] and 13CH313CN [17]. For the first time all the Jo coupling constants except one (2Jo(15N-13C)) have been established for the chemical compound which exhibits strong molecular interactions in the liquid state. The results for acetonitrile are shown in Table 2. As seen, the dependence on density for the 2J(13C-1H) coupling constant is within the experimental error. It certainly means that every value of 2J(13C-1H) measured in the gas phase is very close to the accurate result for an isolated molecule, 2Jo(13C-1H). The other coupling constants of acetonitrile are dependent on the density of gas solvent.
Table 3 gives more experimental coupling constants determined in the gas phase. Some of them were carefully verified over the range of density and found to be independent of density. Such coupling constants can safely be used as the experimental standards (Jo). However, it does not mean that the spin-spin coupling constants presented in Table 3 always remain unchanged in the gas phase. Their values are still dependent on temperature and the sort of gaseous solvent. One can certainly find that every coupling constant from Table 3 is density dependent if the gaseous solvent is changed. In such a case the extrapolated value to the zero-density limit (Jo) will remain the same. For this reason, only the Jo measurements can be recommended as the experimental standards for any comparison with the results of theoretical studies.
Finally, let us demonstrate the benefits of accurate experimental Jo values for quantum-chemistry calculations. As shown in Table 1, the spin-spin coupling constants for 1,2-13C-acetylene are solvent dependent. 1,2-13C-acetylene gives an AA´XX´ NMR spectrum of which the AA´ and XX´ parts have separately been monitored in the gas phase by the 1H and 13C NMR methods [12]. Xenon (Xe) and carbon dioxide (CO2) have been used as gaseous solvents. It enabled us to observe the 1H and 13C NMR spectra of 13C2H2 as a function of Xe (or CO2) density and after extrapolation to zero density it was possible to determine the four spin-spin coupling constants independent of molecular interactions (Jo). Table 4 shows the experimental results together with the theoretical coupling constants for acetylene calculated during last few years.
| Compound | Spin-spin coupling | 1J(NN′) [Hz] | Method, temperature | Reference |
|---|---|---|---|---|
| HD | 2H - 1H | 43.130(15) | 1H, 294 K | cited in ref. [6] |
| CH4 | 13C - 1H | 125.308(20)a | 1H, 300 K | [18] |
| CH4 | 13C - 1H | 125.304(10)a | 13C, 300 K | [18] |
| CH3D | 13C - 1H | 124.953a | 1H, 300 K | [18] |
| CH3D | 13C - 1H | 124.948a | 13C, 300 K | [18] |
| CH3D | 13C - 2H | 19.224a | 13C, 300 K | [18] |
| CHD3 | 13C - 1H | 124.259a | 1H, 300 K | [18] |
| CHD3 | 13C - 1H | 124.262a | 13C, 300 K | [18] |
| CHD3 | 13C - 2H | 19.113a | 13C, 300 K | [18] |
| CD4 | 13C - 2H | 19.056a | 13C, 300 K | [18] |
| CH3Cl | 13C - 1H | 147.5(3) | 1H | [19] |
| (CH3)3N | 13C - 1H | 131.5(2) | 1H | [19] |
| CH3F | 19F - 13C | 160.2(1) | 13C, 298 K | [20] |
| CH2F2 | 19F - 13C | 232.7(1) | 13C, 298 K | [20] |
| CHF3 | 19F - 13C | 272.4(1) | 13C, 298 K | [20] |
| CF4 | 19F - 13C | 259.4(1) | 13C, 298 K | [20] |
| PF3 | 31P - 19F | 1402(1)a | 19F, 298 K | [13] |
| SiH4 | 29Si - 1H | -201.9(1)a | 1H, 298 K | [13] |
| SiH4 | 29Si - 1H | -201.3(4)a | 1H, 298 K | [11] |
| SiHD3 | 29Si - 1H | -199.9(4)a | 1H, 298 K | [11] |
| SF6 | 33S - 19F | 250.1(4)a | 33S, 298 K | [21] |
a Verified and no density dependence was observed within the experimental errors.
Table 4.
Comparison of calculated and observed spin-spin coupling constants [Hz] for 1,2-13C-acetylene at 300 K.
| J0(300K) | Calculated | Observed | ||
|---|---|---|---|---|
| Ref. [22]a | Ref. [7,8]a | Ref. [23]a | Ref.[12] | |
| 1J0(13C-13C) | 170.9 | 179.74 | 174.42 | 174.78 |
| 1J0(13C-1H) | 237.0 | 259.81 | 249.27 | 247.56 |
| 2J0(13C-1H) | 46.4 | 48.02 | 49.38 | 50.14 |
| 3J0(1H-1H) | 9.5 | 10.04 | 9.53 | 9.62 |
The first column of results was obtained by Kaski et al. [22] in 1998, the second set comes the elaborate study performed by Wigglesworth et al. [7,8] in 2000 and the third column presents the calculations done by Jaszuński and Ruud [23] in 2001. An improvement in the accuracy of ab initio calculations of spin-spin coupling is well seen in Table 4 due to the presence of the experimental Jo values for acetylene. It is also true that this comparison is possible because the rovibrational corrections have carefully been calculated by Wigglesworth et al. [7,8]. It is worth noting that the step (1) - (2) in Fig. 1 can be completed only by advanced calculations. NMR measurements of Jo constant as a function of temperature can partially verify the latter calculations but molecules at the equilibrium geometry will never be available for experimental investigations. As seen, experimental and theoretical studies are complementary in the investigations of spin-spin coupling constants for molecules.
Acknowledgement
This work was supported by the Polish State Committee for Scientific Research as the research grant number 4 T09A 035 23 available in years 2002-2005.
References and Notes
- Helgaker, T.; Jaszuński, M.; Ruud, K. Chem. Rev. 1999, 99, 293.
- See for example: Dalton, a molecular electronic structure program, Release 1.2 (2001), Helgaker, T.; Jensen, H.J.Aa.; Jørgensen, P.; Olsen, J.; Ruud, K.; Aagren, H.; Auer, A.A.; Bak, K.L.; Bakken, V.; Christiansen, O.; Coriani, S.; Dahle, P.; Dalskov, E.K.; Enevoldsen, T.; Fernandez, B.; Hättig, C.; Hald, K.; Halkier, A.; Heiberg, H.; Hettema, H.; Jonnson, D.; Kirpekar, S.; Kobayashi, R.; Koch, H.; Mikkelesen, K.V.; Norman, P.; Packer, M.J.; Pedersen, T.B.; Ruden, T.A.; Sanchez, A.; Saue, T.; Sauer, S.P.A.; Schimmelfennig, B.; Sylvester-Hvid, K.O.; Taylor, P.R.; Vahtras, O.
- Malkin, V.G.; Malkina, O.L.; Eriksson, L.A.; Salahub, D.R. The calculation of NMR and EPR spectroscopy parameters using density functional theory in Theoretical and Computational Chemistry vol. 1 Density Functional Calculations; Politzer, P., Seminario, J.M., Eds.; Elsevier: Amsterdam, 1995. [Google Scholar]
- Laszlo, P. Progress in Nuclear Magnetic Resonance Spectroscopy; Emsley, J.W., Feeney, J., Sutcliffe, L.H., Eds.; Pergamon Press: New York, 1967; Chapter 6. [Google Scholar]
- Barfield, M.; Johnston, M.D., Jr. Chem. Rev. 1973, 73, 53. [CrossRef]
- Raynes, W.T.; Panteli, N. Chem. Phys. Lett. 1983, 94, 558.
- Wigglesworth, R. D.; Raynes, W. T.; Kirpekar, S.; Oddershede, J.; Sauer, S. P. A. J. Chem. Phys. 2000, 112, 736.
- Wigglesworth, R. D.; Raynes, W. T.; Kirpekar, S.; Oddershede, J.; Sauer, S. P. A. J. Chem. Phys. 2000, 114, 9192.
- Wigglesworth, R. D.; Raynes, W. T.; Sauer, S. P. A.; Oddershede, J. Mol. Phys. 1998, 94, 851.
- Wigglesworth, R. D.; Raynes, W. T.; Sauer, S. P. A.; Oddershede, J. Mol. Phys. 1997, 92, 77.
- Sauer, S.P.A.; Raynes, W.T.; Nicholls, R.A. J. Chem. Phys. 2001, 115, 5994.
- Jackowski, K.; Wilczek, M.; Pecul, M.; Sadlej, J. J. Phys. Chem. A 2000, 104, 5955. [CrossRef]
- Jameson, A.K.; Reger, J.P. J. Phys. Chem. 1971, 75, 437. [CrossRef]
- Jameson, A.K.; Moyer, J.W.; Jameson, C.J. J. Chem. Phys. 1978, 68, 2873.
- Jackowski, K.; Kubiszewski, M.; Makulski, W. J. Mol. Struct. 2002, 614, 267. [CrossRef]
- Wilczek, M.; Koźmiński, W.; Jackowski, K. Chem. Phys. Lett. 2002, 358, 263.
- Jackowski, K.; Wilczek, M. J. Mol. Struct. in press.
- Bennett, B.; Raynes, W.T.; Anderson, C.W. Spectrochim. Acta 1989, 45A, 821.
- Douglas, A.W.; Dietz, D. J. Chem. Phys. 1967, 46, 1214.
- Jackowski, K.; Raynes, W.T. J. Chem. Res. (S) 1977, 66.
- Jackowski, K.; Wilczek, M.; Makulski, W.; Koźmiński, W. J. Phys. Chem. A 2002, 106, 2829. [CrossRef]
- Kaski, J.; Lantto, P.; Vaara, J.; Jokisaari, J. J. Am. Chem. Soc. 1998, 120, 3993. [CrossRef]
- Jaszuński, M.; Ruud, K. Chem. Phys. Lett. 2001, 336, 473.
© 2003 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.
Share and Cite
MDPI and ACS Style
Jackowski, K. Gas–Phase Studies of Spin–Spin Coupling Constants. Int. J. Mol. Sci. 2003, 4, 135-142. https://doi.org/10.3390/i4030135
AMA Style
Jackowski K. Gas–Phase Studies of Spin–Spin Coupling Constants. International Journal of Molecular Sciences. 2003; 4(3):135-142. https://doi.org/10.3390/i4030135
Chicago/Turabian StyleJackowski, Karol. 2003. "Gas–Phase Studies of Spin–Spin Coupling Constants" International Journal of Molecular Sciences 4, no. 3: 135-142. https://doi.org/10.3390/i4030135