Solvent Effects on Nuclear Magnetic Resonance 2J(C,Hf) and 1J(C,Hf) Spin–Spin Coupling Constants in Acetaldehyde

Abstract
:Introduction

Method of Calculation
Results and Discussion

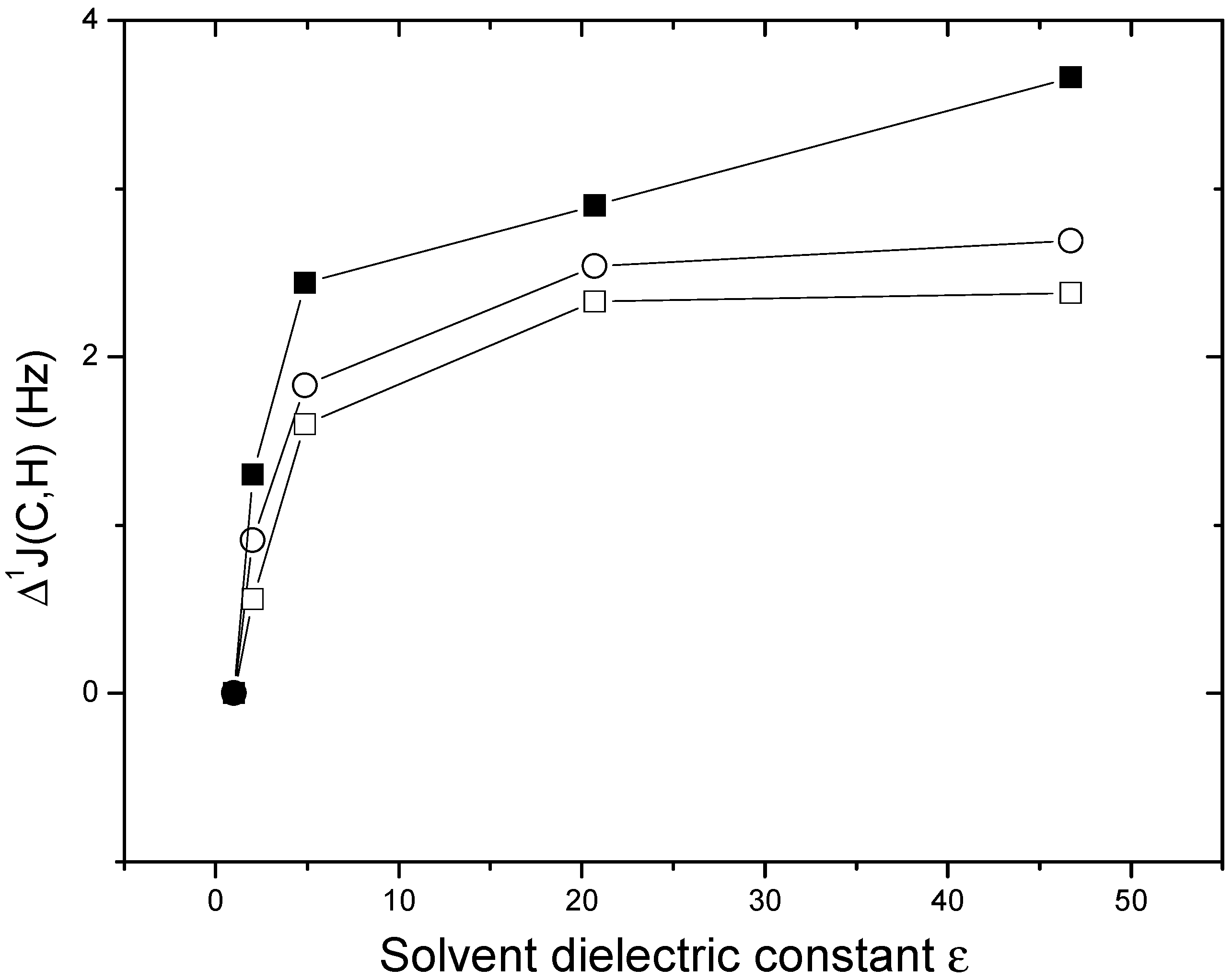{kind=link}
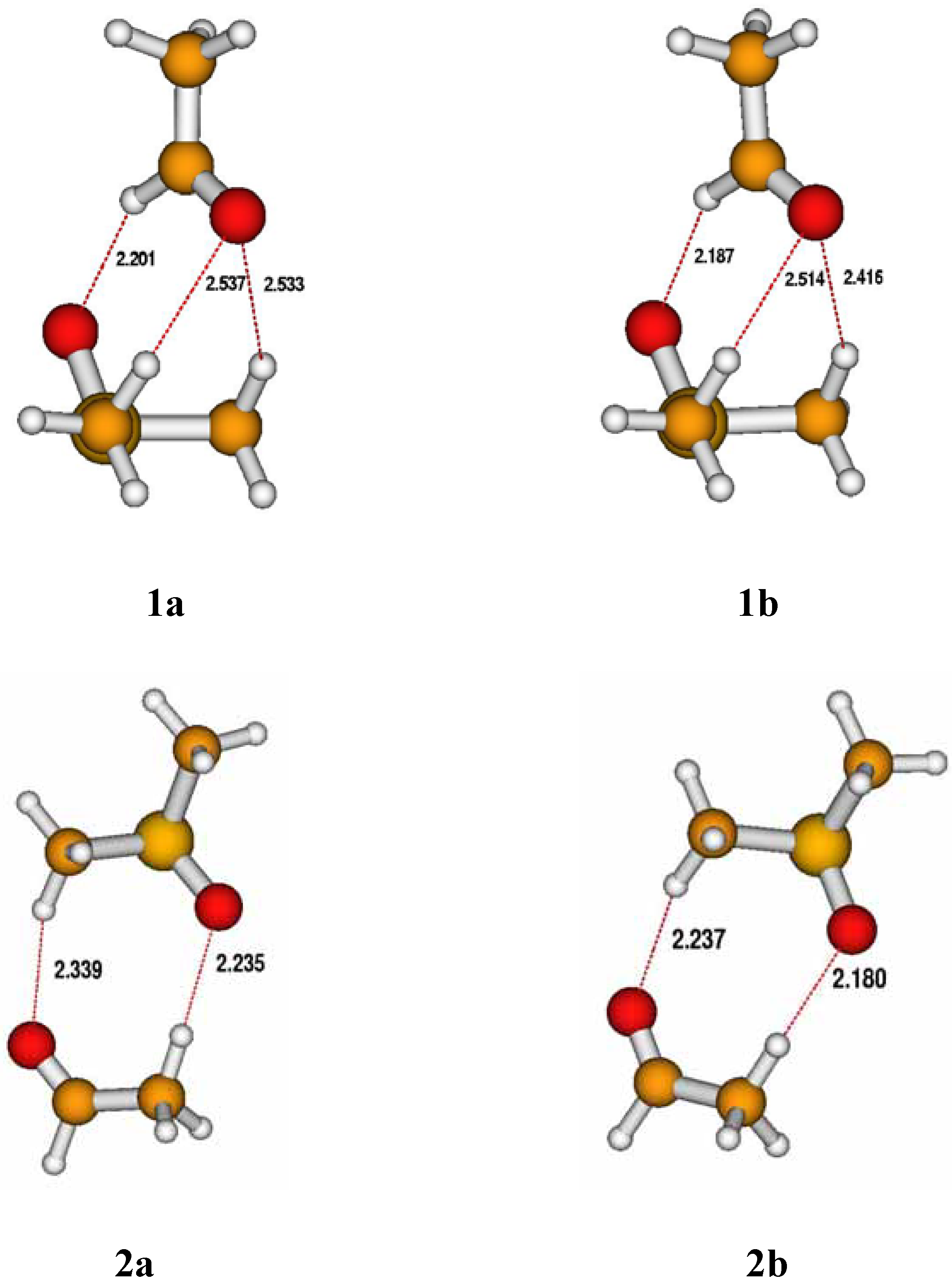{kind=link}
| FC | SD | PSO | DSO | Total | Exp.b) | |
|---|---|---|---|---|---|---|
| 1J(Cc,Hf) (ε = 1) | 177.53 | 0.29 | -0.94 | 0.90 | 177.78 | 169.68 |
| 1J(Cc,Hf) (ε = 46.7) | 179.91 | 0.36 | ncc) | 0.89 | 180.22d) | 173.34 |
| 2J(C1,Hf) (ε = 1) | 32.47 | 0.04 | -0.02 | -0.33 | 32.16 | 29.54 |
| 2J(C1,Hf) (ε = 46.7) | 30.45 | 0.04 | ncc) | -0.34 | 29.95d) | 26.25 |
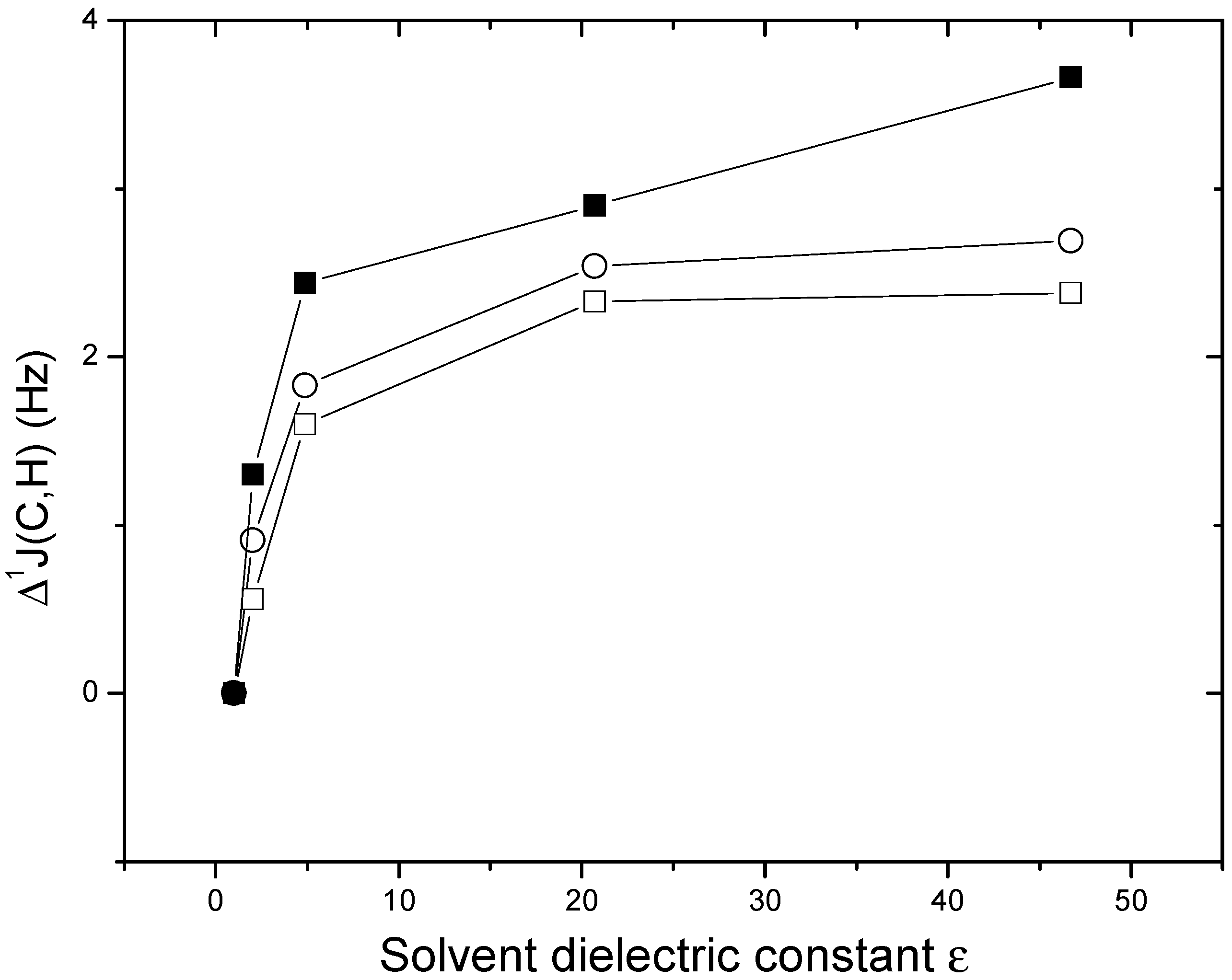


| 1J(Cc,Hf) | 2J(C1,Hf) | |||
|---|---|---|---|---|
| ε = 1b) | ε =46.7c) | ε = 1b) | ε = 46.7c) | |
| Exp. | 169.68 | 173.34 | 29.54 | 26.25 |
| Acet. | 177.53 | 179.91 | 32.47 | 30.45 |
| 1 | 194.09 | 192.79 | 32.25 | 31.15 |
| Δ(1) | 16.56 | 12.88 | -0.22 | 0.70 |
| 2 | 173.36 | 175.99 | 27.37 | 26.31 |
| Δ(2) | -4.17 | -3.72 | -5.20 | -4.14 |
| NLMO | Δ[NLMO] for 1J(Cc,Hf) | Δ[NLMO] for 2J(C1,Hf) | ||
|---|---|---|---|---|
| ε = 1 | ε = 46.7 | ε = 1 | ε = 46.7 | |
| Cc‑C1 | 0.9 | 0.3 | -0.2 | -0.2 |
| Cc‑O | 0.1 | 0.4 | 0.0 | 0.0 |
| Core(C)a) | 1.3 | -0.2 | -0.5 | -0.3 |
| Cc‑Hf | 0.8 | 2.6 | -1.5 | -1.2 |
| CMe—Hb) | -0.1 | 0.0 | 0.0 | 0.3 |
| n(sp) | -0.5 | -0.2 | 0.0 | 0.0 |
| n(p) | 1.5 | 0.7 | 0.6 | -1.6 |
| TOTAL | 4.0 | 3.6 | -1.6 | -3.0 |
| NLMO | Δ[NLMO] for 1J(Cc,Hf) | Δ[NLMO] for 2J(C1,Hf) | ||
|---|---|---|---|---|
| ε = 1 | ε = 46.7 | ε = 1 | ε = 46.7 | |
| Cc‑C1 | 1.0 | 0.4 | -0.3 | -0.4 |
| Core(C) a) | -1.9 | -1.0 | -0.8 | -0.4 |
| Cc‑O | -0.2 | 0.4 | 0.0 | 0.0 |
| CMe—Hb) | 0.0 | 0.1 | 0.1 | 0.3 |
| Cc‑Hf | -5.8 | -3.0 | -1.6 | -1.2 |
| n(sp) | 0.3 | 0.4 | 0.0 | 0.0 |
| n(p) | -1.8 | 0.0 | 0.1 | -1.6 |
| TOTAL | -8.4 | -2.7 | -2.5 | -3.3 |
Concluding Remarks
Acknowledgments
References
- Hansen, P. E. The Chemistry of Double-bonded Functional Groups; Patai, S., Ed.; Wiley, New York, 1989; Ch. 3. [Google Scholar]
- Contreras, R. H.; Peralta, J. E. Angular Dependence of Spin-spin Coupling Constants. Prog NMR Spectrosc. 2000, 37, 321–425. [Google Scholar] [CrossRef]
- Hansen, P. E. Carbon-hydrogen spin-spin coupling constants. Prog. NMR Spectrosc. 1981, 14, 175–295. [Google Scholar] [CrossRef]
- Ando, I.; Inoue, Y.; Watanabe, S.; Sakamoto, Y.; Webb, G. A. The Effects of Solvent Dielectrics on Some NMR Spin-spin Couplings. A Study of Acetaldehyde in the Gaseous Phase and in Various Solvents. J. Mol Liquids 1984, 27, 179–200. [Google Scholar]
- Ando, I. Encyclopedia of Nuclear Magnetic Resonance; Grant, D. M., Harris, R. K., Eds.; Wiley, Chichester, 1996; p. 2512. [Google Scholar]
- Ando, I.; Webb, G. A. Some Quantm Chemical Aspects of Solvent Effects on NMR Parameters. Org. Magn. Reson. 1981, 15, 111–130. [Google Scholar] [CrossRef]
- Ando, I.; Webb, G. A. Theory of NMR Parameters; Academic, New York, 1983. [Google Scholar]
- Ruiz-López, M.; Rinaldi, D.; Rivail, J-L. Electrostatic Solvent Effect on Spin-spin Nuclear Coupling Constants. J. Chem. Res. (M) 1982, 3369–3391. [Google Scholar]
- Facelli, J. C.; Giribet, C. G.; Contreras, R. H. A theoretical studies of medium effects on the transmission mechanisms of the Fermi contact term of spin-spin coupling constants in the acetamide molecule. Int. J. Quantum Chem. 1984, 25, 515–525. [Google Scholar] [CrossRef]
- Helgaker, T.; Jaszunski, M.; Ruud, K. Ab initio Methods for the Calculation of NMR Shielding and Indirect Spin-spin Coupling Constants. Chem. Rev. 1999, 99, 293–352. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Theory and calculation of nuclear spin-spin coupling constants. Prog. NMR. Spectrosc. 1999, 35, 267–294. [Google Scholar] [CrossRef]
- Contreras, R. H.; Peralta, J. E.; Giribet, C. G.; Ruiz de Azúa, M. C.; Facelli, J. C. Advances in Theoretical and Physical Aspects of Spin-spin Coupling Constants. Ann. Rep. NMR Spectrosc. 2000, 41, 55–184. [Google Scholar]
- Åstrand, P-O.; Mikkelsen, K. V.; Jørgensen, P.; Ruud, K.; Helgaker, T. Solvent Effects on Nuclear Shieldings and Spin-spin Couplings of Hydrogen Selenide. J. Chem. Phys. 1998, 108, 2528–2537. [Google Scholar] [CrossRef]
- Mikkelsen, K. V.; Ruud, K.; Helgaker, T. Solvent Effects on the NMR Parameters of H2S and HCN. J. Comput. Chem. 1999, 20, 1281–1291. [Google Scholar] [CrossRef]
- Pecul, M.; Sadlej, J. The nuclear spin-spin coupling constants in the water dimer. Chem. Phys. Lett. 1999, 308, 486–494. [Google Scholar] [CrossRef]
- Malkin, V. G.; Malkina, O. L.; Salahub, D. R. Calculation of spin-spin coupling constants using density functional theory. Chem. Phys. Lett. 1994, 221, 91–99. [Google Scholar] [CrossRef]
- Grayce, C. J.; Harris, R. A. A density functional theory of the Fermi contact contribution to the nuclear spin-spin coupling constant. Chem. Phys. Lett. 1995, 234, 319–322. [Google Scholar] [CrossRef]
- Stahl, M.; Schopfer, U.; Frenking, G.; Hoffmann, R. W. Conformational Analysis with Carbon-Carbon Coupling Constants. A Density Functional and Molecular Mechanics Study. J. Org. Chem. 1997, 62, 3702–3704. [Google Scholar] [CrossRef]
- Dickson, R. M.; Ziegler, T. NMR Spin-Spin Coupling Constants from Density Functional Theory with Slater-Type Basis Functions. J. Phys. Chem. 1996, 100, 5286–5290. [Google Scholar] [CrossRef]
- Cloran, F.; Carmichael, I.; Seriani, A. S. 13C-1H and 13C-13C Spin Coupling Behavior in Aldofuranosyl Rings from Density Functional Theory. J. Phys. Chem. A 1999, 103, 3783–3795. [Google Scholar] [CrossRef]
- Bouř, P.; Buděšínskỷ, M. Sum-over-states Calculation of the Nuclear Spin–spin Coupling Constants. J. Chem. Phys. 1999, 110, 2836–2843. [Google Scholar] [CrossRef]
- Dingley, A. J.; Masse, J. E.; Peterson, R. D.; Barfield, M.; Feigon, J.; Grzesiek, S. Internucleotide Scalar Couplings Across Hydrogen Bonds in Warson-Crick and Hoogsten Base pairs of a DNA Triplex. J. Am. Chem. Soc. 1999, 121, 6019–6027. [Google Scholar] [CrossRef]
- Helgaker, T.; Watson, M.; Handy, N. C. Analytical Calculation of Nuclear Magnetic Resonance Indirect Spin–spin Coupling Constants at the Generalized Gradient Approximation and Hybrid Levels of Density-Functional Theory. J. Chem. Phys. 2000, 113, 9402–9409. [Google Scholar] [CrossRef]
- Sychrovský, V.; Gräfenstein, J.; Cremer, D. Nuclear Magnetic Resonance Spin–spin Coupling Constants from Coupled Perturbed Density Functional Theory. J. Chem. Phys. 2000, 113, 3530–3547. [Google Scholar] [CrossRef]
- Cuevas, G.; Juaristi, E.; Vela, A. Density Functional Calculation of 1JC-H Coupling Constants in Cyclohehane and Diheterocyclohehanes. Repercussion of Stereoelectronic Effects on Couping Constants. J. Phys. Chem. A 1999, 103, 932–937. [Google Scholar] [CrossRef]
- Mertius, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of ab initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Mertius, S.; Tomasi, J. Approximate Evaluations of the Electrostatic Free Energy and Internal Energy Changes in Solution Processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Zaccari, D. G.; Snyder, J. P.; Peralta, J. E.; Taurian, O. E.; Contreras, R. H.; Barone, V. Natural J Coupling (NJC) Analysis of the Electron Lone Pair Effect on NMR Couplings. 2: The Anomeric Effect on 1J(C,H) Couplings and its Dependence on Solvent. Mol. Phys. 2002, 100, 705–715. [Google Scholar] [CrossRef]
- Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, Jr. J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I; Gomperts, R; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M; Replogle, E.S.; Pople, J. A. Gaussian 98, Revision A.7; Gaussian Inc.: Pittsburgh PA, 1998.
- Pople, J. A.; Beveridge, D. L. Approximate Molecular Orbital Theory; McGraw-Hill: New York, 1970. [Google Scholar]
- Peralta, J. E.; Ruiz de Azúa, M. C.; Contreras, R. H. On the Convergence of FPT-DFT Calculations of the Fermi Contact Contribution to NMR Coupling Constants. Theoret. Chem. Acc. 2000, 105, 156–164. [Google Scholar] [CrossRef]
- Peralta, J. E.; Barone, V.; Ruiz de Azúa, M. C.; Contreras, R. H. Finite Perturbation Theory-Density Functional Theory (FPT-DFT) Calculation of the Spin Dipolar Contribution to NMR Spin-spin Coupling Constant. Mol. Phys. 2001, 99, 655–661. [Google Scholar] [CrossRef]
- Helgaker, T.; Jensen, P.; Jørgensen, H. J. A.; Olsen, J.; Ruud, K.; Ågren, H.; Andersen, T.; Bak, K. L.; Bakken, V.; Christiansen, O.; Dahle, P.; Dalskov, E. K.; Enevoldsen, T.; Fernandez, B.; Heiberg, H.; Hettema, H.; Jonsson, D.; Kirpekar, S.; Kobayashi, R.; Koch, H.; Mikkelsen, K. V.; Norman, P.; Packer, M. J.; Saue, T.; Taylor, P. R.; Vahtras, O. Dalton: An Electronic Structure Program; Release 1.0; 1997. [Google Scholar]
- Guilleme, J.; San Fabián, J. Basis Sets and Active Space in Multiconfigurational Self-Consistent Field Calculations of Nuclear Magnetic Resonance Spin–spin Coupling Constants. J. Chem. Phys. 1998, 109, 8168–8181. [Google Scholar] [CrossRef]
- Peralta, J. E.; Contreras, R. H.; Snyder; J. P. Natural Bond Orbital Disecction of Fluorine-Fluorine Through-space NMR Coupling (JF,F) in Polycyclic Organic Molecules. J. Chem. Soc. Chem. Commun. 2000, 20, 2025–2026. [Google Scholar] [CrossRef]
- Barone, V.; Peralta, J. E.; Contreras, R. H.; Sosnin, A. V.; Krivdin, L. B. Natural J Coupling (NJC) Analysis of the Electron Lone Pair Effect on NMR Couplings: Part 1. The Lone Pair Orientation Effect of an -Nitrogen Atom on 1J(C,C) Couplings. Magn. Reson. Chem. 2001, 39, 600–606. [Google Scholar] [CrossRef]
- Reed, A. E.; Curtiss, L. A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E. D.; Reed, A. E.; Carpenter, J. E.; Weinhold, F. NBO version 3.1, (Included in the Gaussian 98 package of programs).
- Boys, S. F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures With Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Turi, L.; Dannenberg, J. J. Molecular Orbital Studies of C-H...O Hydrogen-bonded Complexes. J. Phys. Chem. 1993, 97, 7899. [Google Scholar] [CrossRef]
- Desiraju, G. R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, 1999. [Google Scholar]
- Kowalewski, D. G.; Kowalewski, V. J.; Peralta, J. E.; Eskuche, G.; Contreras, R. H.; Esteban, A. L.; Galache, M. P.; Díez, E. Intramolecular Electric Field Effect on a 1J(C,H) NMR Spin-spin Coupling Constant. an Experimental and Theoretical Study. Magn. Reson. Chem. 1999, 37, 227–231. [Google Scholar] [CrossRef]
- Vizioli, C.; Ruiz de Azúa, M. C.; Giribet, C. G.; Contreras, R. H.; Turi, L.; Dannenberg, J. J.; Rae, I. D.; Weigold, J. A.; Malagoli, M.; Zanasi, R.; Lazzeretti, P. Proximity Effects on Nuclear Spin-spin Coupling Constants. 1. 1J(CH) Couplings in the Vicinity of an Atom Bearing Lone Pairs. J. Phys. Chem. 1994, 98, 8858–8861. [Google Scholar] [CrossRef]
- Giribet, C. G.; Vizioli, C. V.; Ruiz de Azúa, M. C.; Contreras, R. H.; Dannenberg, J. J.; and Masunov, A. Proximity Effects on Nuclear Spin-spin Coupling Constants. Part 2. The Electric Field Effect on 1J(CH) Couplings. J. Chem. Soc. Faraday Trans. 1996, 92, 3029–3030. [Google Scholar] [CrossRef]
- Taha, A. N.; True, N. S. Experimental 1H NMR and Computational Studies of Internal Rotation of Solvated Formamide. J. Phys. Chem, A 2000, 104, 2985–2993. [Google Scholar] [CrossRef]
- Wigglesworth, R. D.; Raynes, W. T.; Kirpekar, S.; Oddershede, J.; Sauer, S. P. A. Nuclear spin-spin coupling in the acetylene isotopomers calculated from ab initio correlated surfaces for 1J(C,H), 1J(C,C), 2J(C,H) and 3J(H,H). J. Chem. Phys. 2000, 112, 3735–3746. [Google Scholar] [CrossRef]
© 2003 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.
Share and Cite
Zaccari, D.; Barone, V.; Peralta, J.E.; Contreras, R.H.; Taurian, O.E.; Díez, E.; Esteban, A. Solvent Effects on Nuclear Magnetic Resonance 2J(C,Hf) and 1J(C,Hf) Spin–Spin Coupling Constants in Acetaldehyde. Int. J. Mol. Sci. 2003, 4, 93-106. https://doi.org/10.3390/i4030093
Zaccari D, Barone V, Peralta JE, Contreras RH, Taurian OE, Díez E, Esteban A. Solvent Effects on Nuclear Magnetic Resonance 2J(C,Hf) and 1J(C,Hf) Spin–Spin Coupling Constants in Acetaldehyde. International Journal of Molecular Sciences. 2003; 4(3):93-106. https://doi.org/10.3390/i4030093
Chicago/Turabian StyleZaccari, Daniel, Verónica Barone, Juan E. Peralta, Rubén H. Contreras, Oscar E. Taurian, Ernesto Díez, and Angel Esteban. 2003. "Solvent Effects on Nuclear Magnetic Resonance 2J(C,Hf) and 1J(C,Hf) Spin–Spin Coupling Constants in Acetaldehyde" International Journal of Molecular Sciences 4, no. 3: 93-106. https://doi.org/10.3390/i4030093