Ab initio Study of Alkyl-oxonium Cations CnH2n+1OH2+, n=1,2,3,4

Laboratoire de Mathématiques Appliquées (L.M.A), E.N.S.Techniques Avancées 32 Boulevard Victor 75739 Paris Cedex, France
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2004, 5(4), 110-118; https://doi.org/10.3390/i5040110
Submission received: 26 November 2003
/
Accepted: 5 July 2004
/
Published: 8 August 2004
Abstract
:Within the framework of the itinerant radical model, the solvated electron in liquid alcohols is understood as an itinerant alkyl-oxonium ROH2. radical. As a first step in the investigation of those radicals, this study deals with the optimization of related ROH2+ alky-oxonium cations: CnH2n+1OH2+,n=1,2,3,4. The structures were optimized at the MP2/6-31G**++ level with the help of the GAMESS ab initio package. Optimized structures are reported for the following cations: MethylOxonium, EthylOxonium, 1-PropylOxonium, 2-PropylOxonium, 1-ButylOxonium, 2-ButylOxonium, IsoButylOxonium, and TertButylOxonium. Optimized geometries are displayed with the help of the ChemApp Java applet. Vibrational frequencies and ZPEs have been computed, and visual depictions of expected experimental IR spectra have been simulated with the help of Lorentzian functions.
Introduction
According to the itinerant radical model [1],[2],[3], the entity that is currently called a hydrated electron or (e-)aq should be understood instead as the mobile H3O. protic defect or itinerant hydronium radical. In a way similar to the F-center, also a neutral defect, the hydronium radical neutral defect moves under the influence of an electric field in the same direction as a negatively charged classical species.
Similarly, in alcohols, the solvated electron (e-)solv should be understood as the itinerant alkyl-oxonium ROH2. radical. This conception would help to understand why the absorption spectrum of the solvated electron (e-)solv appears to depend on chemical properties, instead of physical properties such as the dielectric constant as predicted by the cavity model. Furthermore, Gordon Freeman and co-workers [4] discovered that the mobility of (e-)solv is closely related to the mobility of ROH- in various alcohols, a fact which is very hard to understand within the framework of the cavity model. Within the itinerant radical model, on the contrary, it is expected that mobilities of ROH- and ROH2. are related since both mobility mechanisms involve motion of itinerant protic defects.
So far to our knowledge, the structures and dynamics of positive protic defects ie ROH2+ in liquid alcohols have been theoretically studied only in liquid methanol by Mark Tuckerman and coworkers [5].
With the help of ab initio methods, it is not currently feasible to compute the absorption spectra of solvated alkyl-oxonium radicals. However we believe that the most important factor, that influences the absorption spectrum of various ROH2. radical, is their respective intramolecular structures. Therefore, our ultimate goal is to compute the absorption spectra of a significant number of isolated alkyl-oxonium radicals. Then, we expect that computed isolated alkyl-oxonium radical respective spectral shifts should match with observed respective spectral shifts between "solvated electrons" in various alcohols.
As a first step towards determining of the absorption spectra of isolated ROH- in various alcohols in order to check the prediction of the itinerant radical model with experimental spectra, we have focused this study in determining the optimized geometries of the isolated ROH2+ alkyl-oxonium cations: CnH2n+1OH2+,n=1,2,3,4.
Molecular structures were optimized at the MP2/6-31G**++ level with the help of the ab initio package GAMESS (General Atomic and Molecular Electronic Structure System) [6]. The computations were performed on clusters of Linux PC machines at ENSTA. Geometry optimizations were performed for the following cations: MethylOxonium; EthylOxonium; 1-PropylOxonium, 2-PropylOxonium and 1-ButylOxonium, 2-ButylOxonium, IsoButylOxonium, TertButylOxonium. Vibrational frequencies and ZPEs are computed, and visual depictions of expected experimental IR spectra are simulated with the help of Lorentzian functions. MP2 Frequencies were not scaled. Curves are displayed with the help of the Xgraphic software [7].
With the help of the graphics package MolDen, edition of Z-matrices and production of 2D images were performed [8].
Since static 2D images are not often providing an easy way to understand molecular structures, optimized geometries are also displayed ( see HTML version of this paper ) with the help of the freely available ChemApp Java applet [9] written by John and George Purvis. This did not appear obvious to us when our graphics were first created during the summer of 1999, but the CAChe format is now linked to a proprietary product distributed by Cache Software [10], a Fujitsu subsidiary. Most regretfully, the original ChemApp internet page no longer exists. We have nevertheless preserved an almost complete copy of the original site that we have made available to our readers for convenience. This did not appear obvious to us when our graphics were first created during the summer of 1999, but the CAChe format is now linked to a proprietary product distributed by Cache Software [10], a Fujitsu subsidiary. Since the ChemApp applet [9] reads only files in the CAChe format [11], we used the program Babel [12] (A Molecular Structure Information Interchange Hub ) to convert GAMESS outputs into CAChe files. There is now a Open Source successor to Babel : OpenBabel [13] . When the original ChemApp site was still online, Dr. George Purvis provided us a very kind help in order to diagnose a bug and find a way to avoid it. It is also interesting to notice that the CAChe format [11] allows also to display Molecular Orbitals This feature has not been used in this article.
The HTML version of this paper contains many pictures, data and supplementary material that could not be reasonably provided in a printer-ready PDF format.
Determination of Initial Geometries
A first step is to determine initial reasonnable geometries that are going to be included in the GAMESS input file, before starting energy computation and geometry optimization. We have used the geometry database server CORINA (COoRdINAtes) [14] that generates a set of atomic coordinates. In order to use CORINA [14] one should enter the description of the molecule using a SMILES ( Simplified Molecular Input Line Entry Specification ) string [15]. The SMILES [15] strings concerning alkyl-oxonium cations under study are listed below ( cf. Table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Formulae | SMILES string |
|---|---|---|
| MethylOxonium Cation | CH3-OH2+ | C[OH2+] |
| EthylOxonium Cation | CH3-CH2-OH2+ | CC[OH2+] |
| 1-PropylOxonium Cation | CH3-CH2-CH2-OH2+ | CCC[OH2+] |
| 2-PropylOxonium Cation | CH3-CH(OH2+)-CH3 | C(C)(C)[OH2+] |
| 1-ButylOxonium Cation | CH3-CH2-CH2-CH2-OH2 + | CCCC[OH2+] |
| 2-ButylOxonium Cation | CH3-CH(OH2+)-CH2-CH3 | CC(CC)[OH2+] |
| IsoButylOxonium Cation | (CH3)2-CH-CH2-OH2+ | C(C)(C)C[OH2+] |
| TertButylOxonium Cation | (CH3)3-C-OH2+ | C(C)(C)(C)[OH2+] |
Computations of Optimized Geometries
Concerning the MethylOxonium Cation, a symmetric Cs geometry seems a rather obvious choice. We nevertheless optimized with and without a Cs symmetry constraint, as expected the geometry and energy are the same. The vibronic energy is the sum of the electronic energy and the vibrational Zero-Point-Energy (ZPE), computed within the framework of the adiabatic approximation and the harmonic approximation (cf. Table 2). The fact that the ZPE is very slightly lower in magnitude in the Cs geometry is just an artefact.
| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) |
|---|---|---|
| C1 CORINA | -115,693.616 | -115.627,745 |
| Cs CORINA | -115,693.616 | -115.627,746 |
Concerning the EthylOxonium Cation, we started from the structure proposed by CORINA which appears as a reasonable guess ( cf. Table 3 ). The ZPE is 0.094,880 Hartree.
| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) |
|---|---|---|
| C1 CORINA | -154.889,501 | -154,794,621 |
Concerning the 1-PropylOxonium Cation, we started from the structure proposed by CORINA without symmetry constraint. The ZPE is 0,124.270 Hartree. The initial and final geometries seem quite reasonable, and no other geometries were investigated ( cf. Table 4 ).
| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) |
|---|---|---|
| C1 CORINA | -194.074,869 | -194.049,401 |
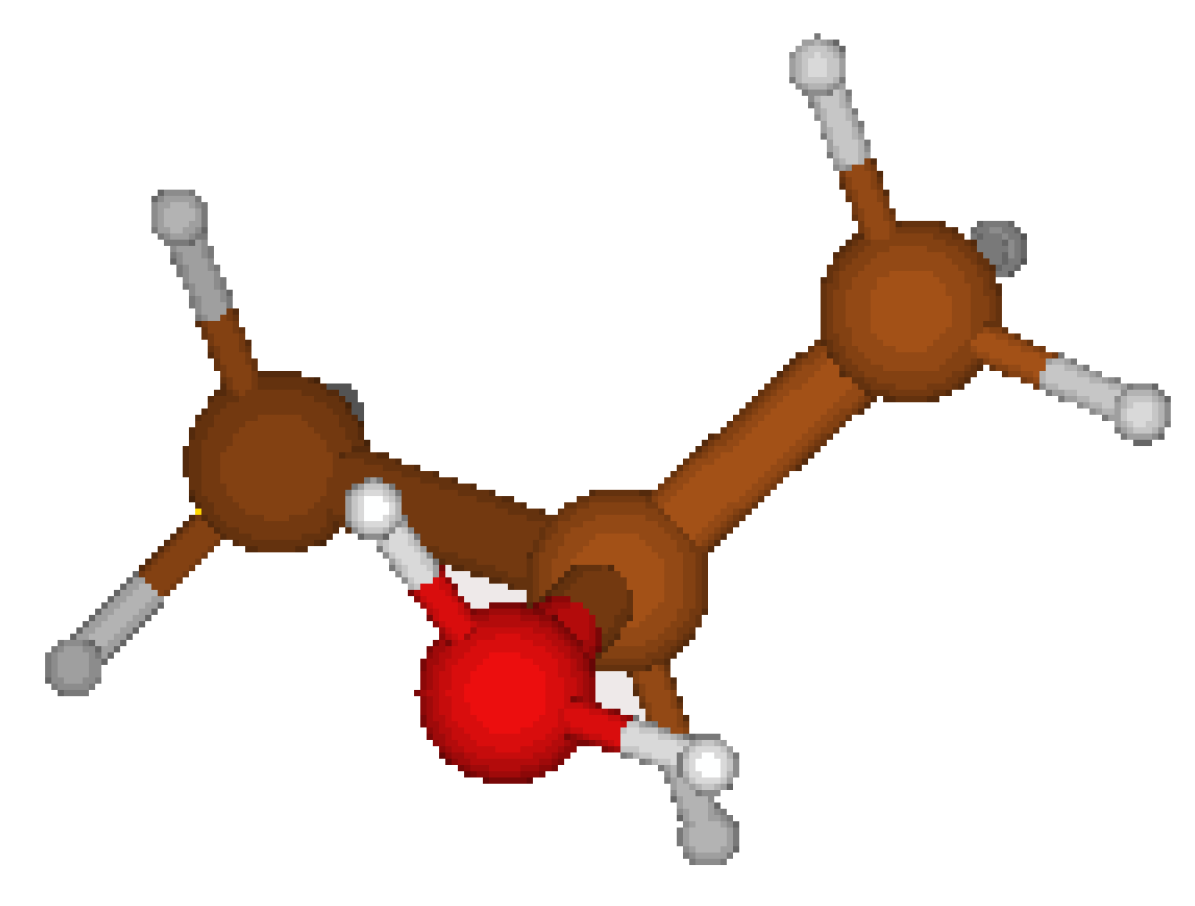
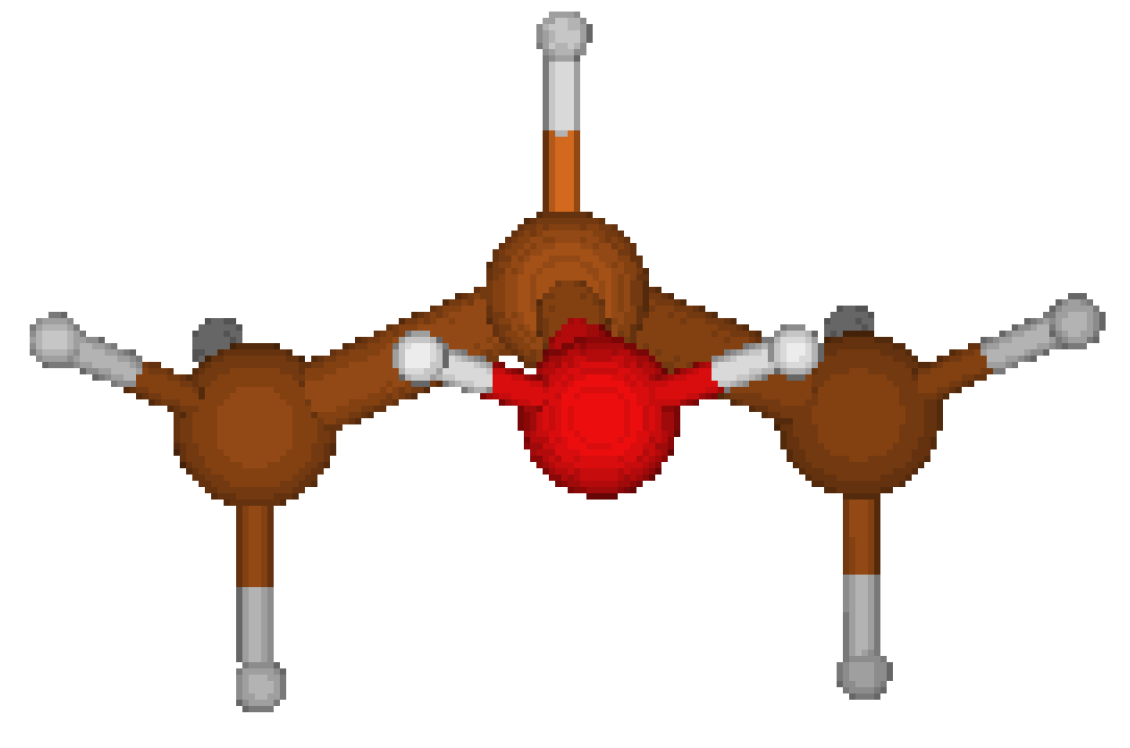
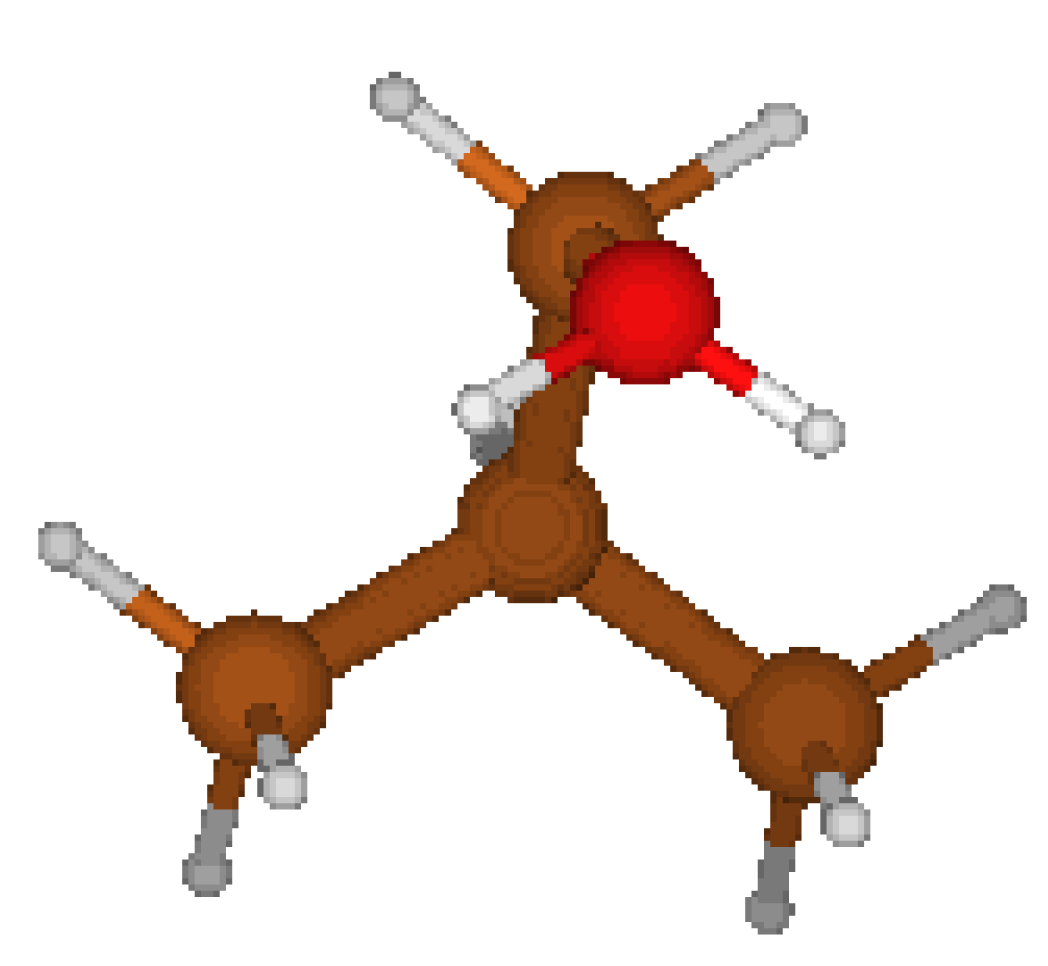
Concerning the 2-PropylOxonium Cation, we started from the structure proposed by CORINA which is asymmetric (cf. Fig 1) . We tried yet another geometry (cf. Fig 2) where hydrogen atoms nearby the oxygen atoms are located respectively on each side of a symmetry plane ( cf. Table 5). This Cs structure features a lower energy. It appears that CORINA failed to guess the optimized geometry in this case.
Figure 1.
C1 structure CORINA.

Figure 2.
Cs structure.

| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) |
|---|---|---|
| C1 CORINA | -194,085.307 | -193.962,213 |
| Cs | -194,085.994 | -193,962.813 |
1-ButylOxonium Cation: We kept the structure proposed by CORINA. It seems difficult to envision any other reasonable candidate geometry. Trying to impose an overall Cs symmetry would require two pair of hydrogen atoms to face one another.
| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) |
|---|---|---|
| C1 CORINA | -233.259,678 | -233.106,238 |
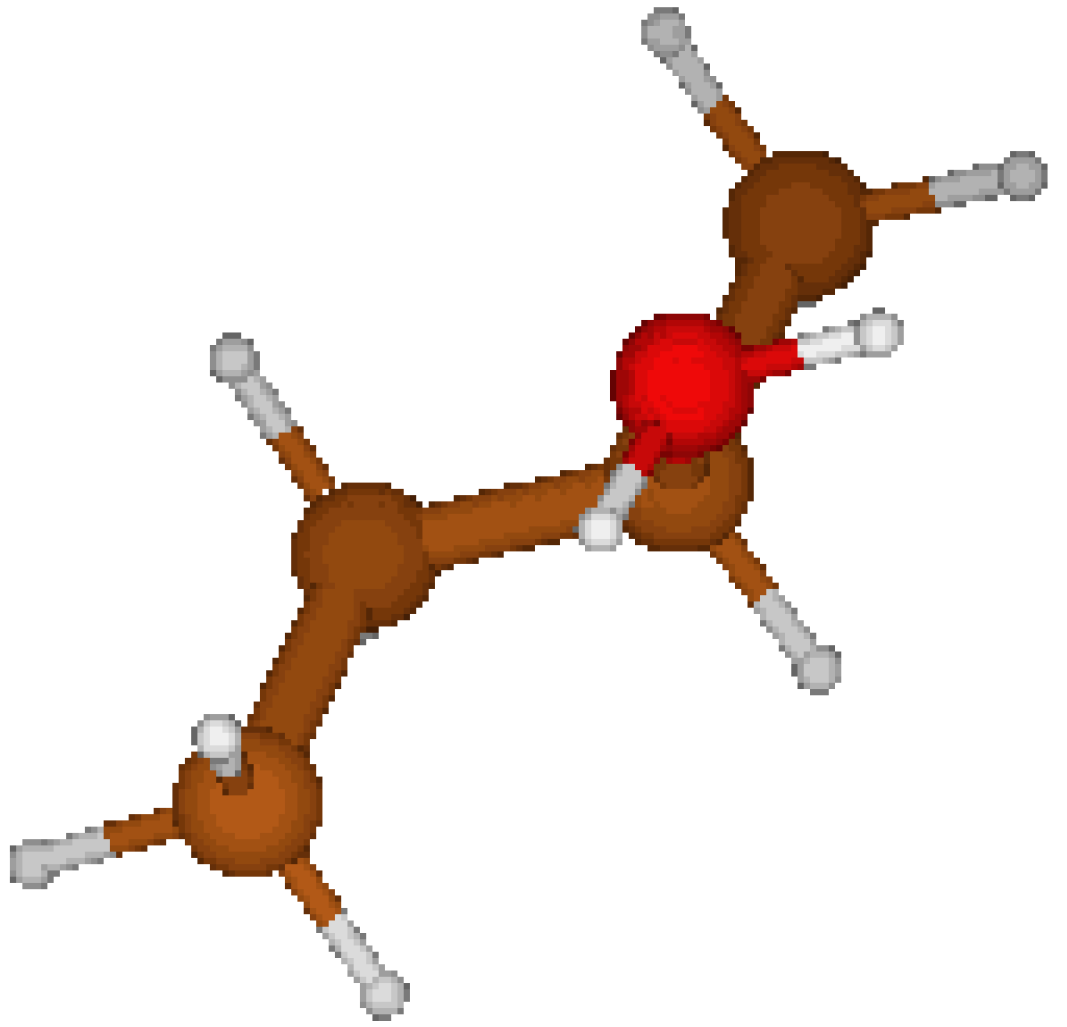
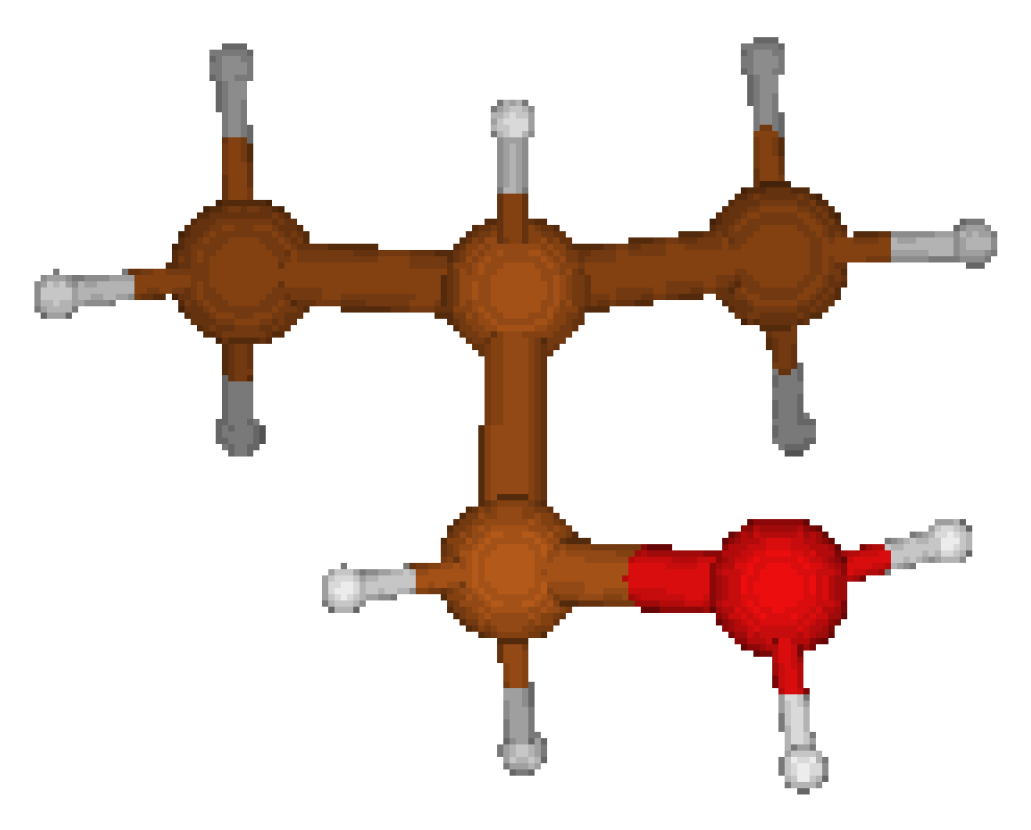
2-ButylOxonium Cation: We considered two types of geometries : the first one being proposed by CORINA, the second one is obtained from the former geometry by a 120° rotation of the oxonium group. This latter geometry ( nearly symmetric in the vicinity of the oxonium group) features a lower energy (cf. Table 7). It appears that CORINA again failed to guess the optimized geometry.
Figure 3.
C1 structure CORINA.

Figure 4.
nearly Cs structure.

| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) | Nature |
|---|---|---|---|
| C1 CORINA | -233,271.199 | -233,118.970 | local minimum |
| nearly Cs | -233,271.777 | -233,119.432 | global minimum |
IsoButylOxonium Cation : We considered four types of geometry. The first one is an asymmetric geometry proposed by CORINA ( cf Fig 5 ). Since this molecule, at a very first glance, could feature a symmetry plane, we explored also three other more symmetric Cs geometries, among which one ( cf Fig 6 type B) turned to be a local minimum. The geometry proposed by CORINA however prevailed as the best geometry for a global minimum (cf Table 8).
Figure 5.
C1 CORINA.

Figure 6.
Cstype B.

| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) | Nature |
|---|---|---|---|
| C1 CORINA | -233.263,333 | -233.110,484 | global minimum |
| Cs type A | -233.255,232 | -233.102,368 | saddle point |
| Cs type B | -233.262,854 | -233.110,094 | local minimum |
| Cs type C | -233.260,681 | -233.108,464 | saddle point |
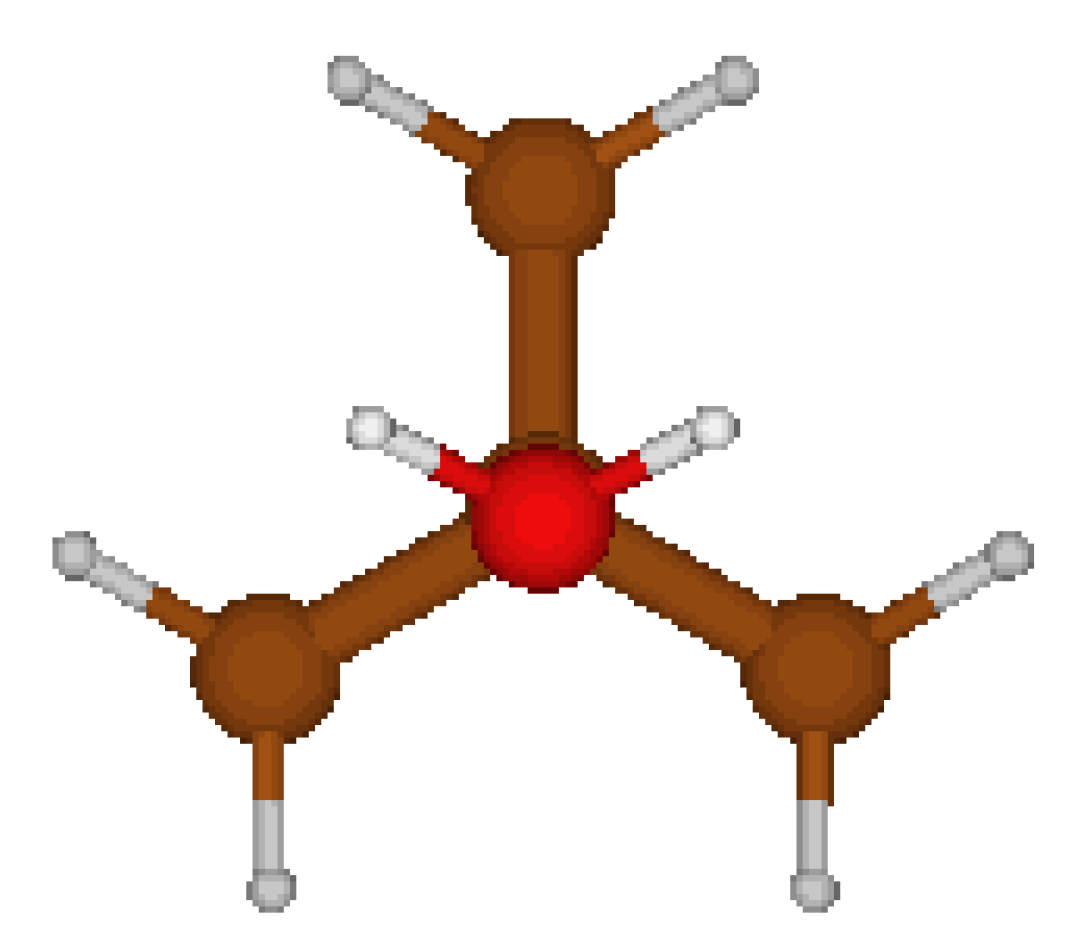
TertButylOxonium Cation : CORINA suggested a very reasonable Cs (cf Fig 7) symmetric structure. We also explored another Cs symmetric structure which turned to be, as expected, a saddle point (cf. Table 9).
| Symmetry | Electronic Energy (Hartree) | Vibronic Energy (Hartree) | Nature |
|---|---|---|---|
| Cs CORINA | -233.282,026 | -233.130,937 | global minimum |
| Cs | -233.280,442 | -233.129,881 | saddle point |
Figure 7.
Cs global minimum.

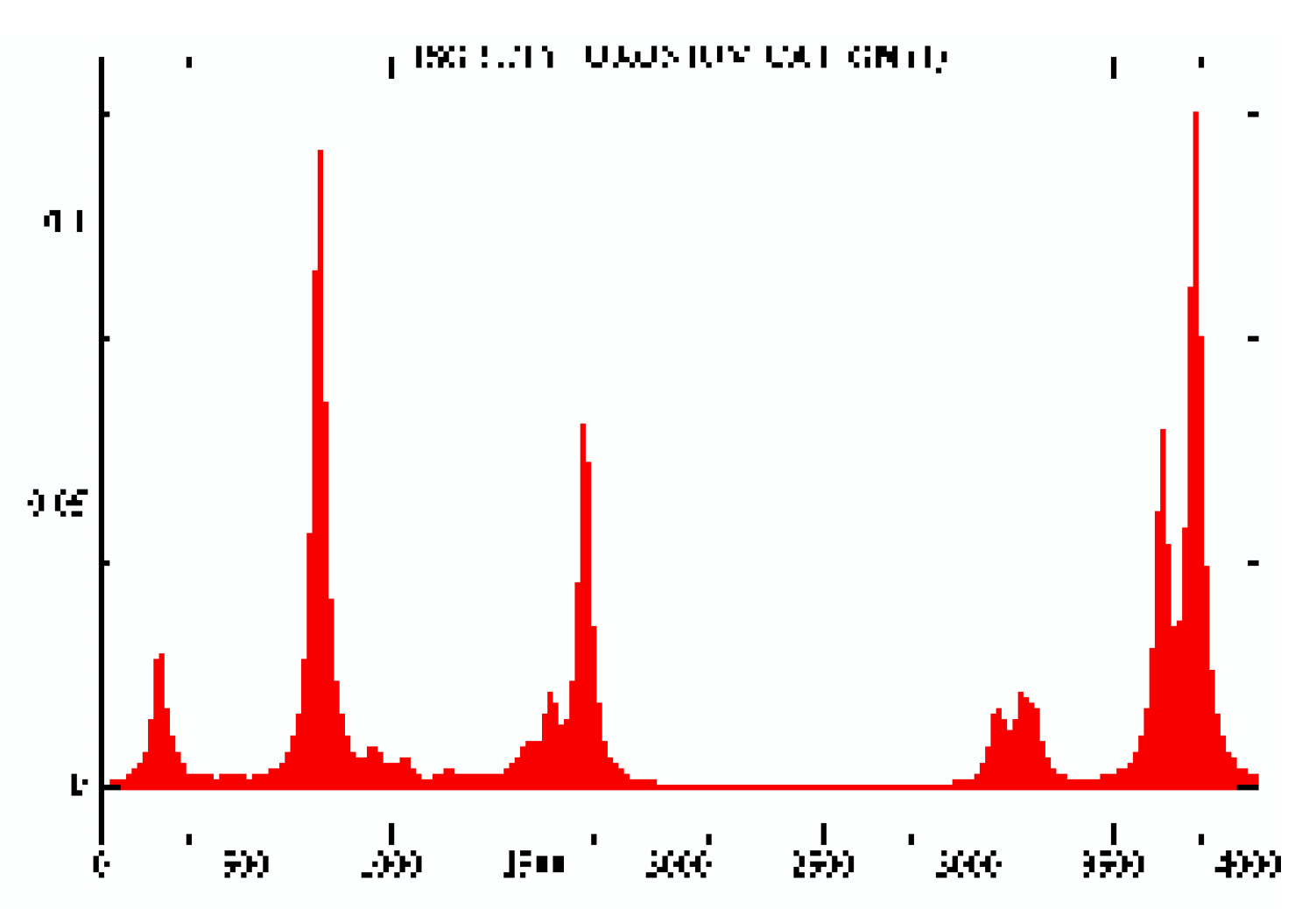
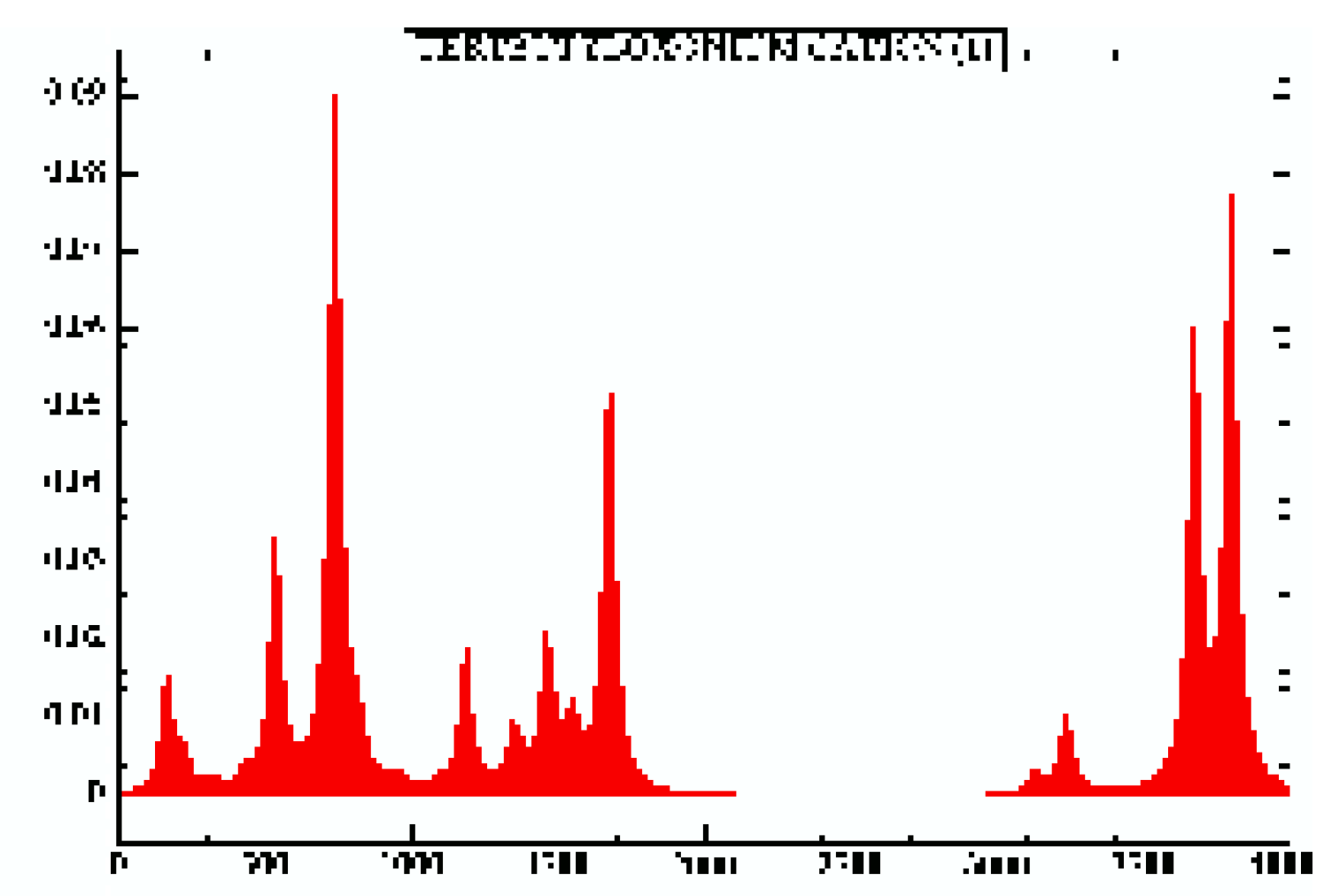
Vibrational absorption spectra are also reported for each geometry under consideration and could provide some help to identify molecular geometries, for example the IsoButylOxonium global minimum (cf Fig. 8 ) and the TertButylOxonium global minimum (cf. Fig. 9). For more see the HTML version.
Figure 8.
IsoButylOxonium (Y intensity au / X frequency cm-1).

Figure 9.
TertButylOxonium.

Discussion
Costly Hessian computations were achieved for three purposes : first to determine the topological nature of geometries on the Potential Energy Surface, second to take into account the ZPE, within the adiabatic and harmonic approximations, and third to compute the vibrational spectra.
We took care of including ZPE contributions in determining the lower geometry for each species. However, ZPE contributions did not modify the choice between stationary points that were under consideration, among possible candidates for the global minimum geometry. The lowest electronic energy geometry turned always always to be the lowest vibronic energy. No dramatic isotopic substitution effects are therefore envisioned, at least in the gas phase.
The geometry predictions obtained from CORINA are, in general, validated by our MP2 computations. However, in the two cases of 2-PropylOxonium and 2-ButylOxonium, do CORINA predictions appear to be in disagreement with our ab initio computations. This corresponds to a 75% success rate.
The fact that that the IsoButylOxonium cation does not feature any symmetry is going to unfortunately imply more costly computations in order to estimate the electronic absorption spectrum of the corresponding radical.
Interactive techniques of geometry visualization, such as provided by the ChemApp Java applet allow to understand and investigate geometry structures in a much more convenient way than 2D static pictures. One big advantage of a Java applet is that it does not require the reader to have any helper application or plugin installed in her/his browser. However, many browsers have the Java compatiblity disabled, and it take times to load the Java interpreter into memory. Once the Java Virtual Machine (JVM) is loaded, other visualizations are much quicker to appear.
Another aspect concerning visualization tools is that a freely available software does not necessarily means that it is a free software under the GPL [16] or an open source software. At the time, in 1999, when our decision was made, we were not conscious enough of the issues involved in the long term. There is no guarantee that a software that was freely available in 1999 will continue to be easily available or updated. This is a lesson that has been learned.
Conclusion
We computed the lower energy geometries and infrared absorption spectra of the isolated small CnH2n+1OH2+,n=1,2,3,4 alkyl-oxonium cations : MethylOxonium, EthylOxonium, 1-PropylOxonium, 2-PropylOxonium, 1-ButylOxonium , 2-ButylOxonium , IsoButylOxonium and TertButylOxonium, while taking into account the ZPE.
The geometrical structures proposed by CORINA [14] appeared to be the correct guesses in six out of eight cases. The two prediction failures concern the 2-PropylOxonium and 2-ButylOxonium Cations.
Vibrational absorption spectra are also reported for each geometry under consideration.
The usefulness of a freely available Java Applet has been demonstrated in order to communicate research results concerning molecular structures. However, a freely available software is not necessarily a free software or an open source software.
Acknowledgements
We deeply thank Professor Robert Topper for organizing the ECCC9 conference. Dr. George Purvis [11] , coauthor of ChemApp Java applet [9] is thanked for his help in order to find a way to avoid a bug originating from BABEL [12] or ChemApp [9]. Fréderic Haitayan, performed this research project as an ENSTA student within the framework of his Personal Project in a Laboratory ( PPL ).This work has been performed with the specific computer and network support of ENS Techniques Avancées (E.N.S.T.A).
References and Notes
- Muguet, F. F. Investigation of diffuse intermolecular electronic systems. Ph.D. dissertation, (Texas Tech University, 1992) Publisher, UMI (Ann Arbor, MI, USA).
- Muguet, F. F.; Gelabert, H.; Gauduel, Y. MCSCF-RPA study of the electronic excitation spectrum of the hydrated hydronium radical. J. Chim. Phys. 1996, 93, 1808–1827. [Google Scholar]
- Muguet, F. F. Coupling between Molecular Orbital Shape Evolution and Inversion Vibration within the Hydronium Radical. Internet J. Chem. 1998, 1, 25. [Google Scholar]
- Zhao, Y.; Freeman, G. R. Solvated electron mobility in liquid tert-butanol. Can. J. Chem. 1995, 73, 389–391. [Google Scholar] [CrossRef]
- Morrone, J. A.; Tuckerman, M. E. Ab initio molecular dynamics study of proton mobility in liquid methanol. J. Chem. Phys. 2002, 117, 4403. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldrige, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; Windus, T.L.; Dupuis, M.; Montgomery, J.A. J. Comput. Chem.; 1993; 14, pp. 1347–1363, GAMESS. (http://www.msg.ameslab.gov/GAMESS/GAMESS.html). [CrossRef]
- Jouve, F. Xgraphic Version 4.04. Graphic software. (http://www.cmap.polytechnique.fr/XGRAPHICS/xgraphics.html).
- Schaftenaar, G. Molden graphics package. (http://www.caos.kun.nl/~schaft/molden/molden.html).
- Purvis, J.; Purvis, G. ChemApplet, a Java Chemical Sample 3D Molecule Structure Viewer (JARS, Oct. 18 1998). Preserved at http://www.cyberchemistry.com/ChemApp/index.html.
- CAChe Group, Fujitsu America, Inc. (http://www.cachesoftware.com).
- Purvis, G. D. The Chemical Sample: A Fundamental Object for Molecular Modeling. J. Chem. Inf. Comp. Sci. 1994; 34, 17–21. [Google Scholar]
- Walters, P.; Stahl, M. Babel - A Molecular Structure Information Interchange Hub. (http://smog.com/chem/babel).
- Open Babel. (http://openbabel.sourceforge.net/).
- CORINA (CooRdINAtes), Fast and Efficient Generation of High-Quality 3D Molecular Models. (http://www2.chemie.uni-erlangen.de/software/corina/index.html).
- SMILES, Simplified Molecular Input Line Entry Specification. (http://www.daylight.com/dayhtml/smiles/index.html).
- GPL, General Public License. (http://www.gnu.org/copyleft/gpl.html).
© 2004 by MDPI (http://www.mdpi.org). Reproduction for noncommercial purposes permitted.
Share and Cite
MDPI and ACS Style
Haitayan, F.; Muguet, F.F. Ab initio Study of Alkyl-oxonium Cations CnH2n+1OH2+, n=1,2,3,4. Int. J. Mol. Sci. 2004, 5, 110-118. https://doi.org/10.3390/i5040110
AMA Style
Haitayan F, Muguet FF. Ab initio Study of Alkyl-oxonium Cations CnH2n+1OH2+, n=1,2,3,4. International Journal of Molecular Sciences. 2004; 5(4):110-118. https://doi.org/10.3390/i5040110
Chicago/Turabian StyleHaitayan, Frédéric, and Francis F. Muguet. 2004. "Ab initio Study of Alkyl-oxonium Cations CnH2n+1OH2+, n=1,2,3,4" International Journal of Molecular Sciences 5, no. 4: 110-118. https://doi.org/10.3390/i5040110