Study of Polymorph Prediction For L-Ascorbic Acid

1
Department of Chemistry and Chemical Biology, Stevens Institute of Technology, Hoboken, New Jersey 07030, USA
2
Highly Filled Materials Institute (HFMI), Stevens Institute of Technology, Hoboken, New Jersey 07030, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2005, 6(12), 291-302; https://doi.org/10.3390/i6120291
Submission received: 31 October 2005
/
Accepted: 29 November 2005
/
Published: 6 December 2005
Abstract
:Possible polymorphs of L-ascorbic acid were investigated, considering eight space groups and assuming one molecule in the asymmetric unit. The grid-search method was compared with a Monte Carlo approach as performed in the Biosym / MSI polymorph Predictor. A number of possible crystal structures were found, including the experimental structure. Energy minimizations were performed with a united-atom force field. In all cases, the experimental structure had a low lattice energy. The number of hypothetical crystal structures was reduced considerably by removing space-group symmetry constraints, or by a primitive molecular dynamic shake-up. Nevertheless, sufficient structures of equal or lower energy compared with the experimental structure remained to suggest that other factors need to be considered for polymorph predictions of materials.
1. Introduction
The relationship between the crystal morphology and the internal arrangement of atoms in the crystal is of great interest to chemists and chemical and process engineers. If the drug can crystallize in two or more polymorphs with different bioavailabilities, the optimal dose depends on the polymorph used in the formulation. The polymorphic behavior of organic and inorganic solids can be of crucial importance in the pharmaceutical industry [1]. Properties varying between polymorphs include stability (i.e., shelf life of drug), crystal shape, compressibility, density (important in formulation), and the dissolution rate (important in determining bioavailability). Polymorphism will affect these products during downstream development and formulation [2]. The existence of different crystal forms impacts key properties such as shelf life, vapor pressure, solubility, bioavailability, morphology, density and shock sensitivity. It is vital that researchers involved in the formulation of crystalline products have the ability to select the polymorph with the correct properties and anticipate problems such as the unwanted crystallization of other polymorphs. In order to do this they need to establish likely polymorphic forms. This knowledge is also important for patenting and registration purposes. It is usually impossible or impractical to use single crystal X-ray diffraction. To find the possible polymorph in which a given molecule can crystallize the researcher often needs to piece together experimental results such as the powder diffraction patterns and lattice energy calculations [3]. Polymorphism can cause profound problems in the development and formulation of organic and inorganic crystalline products. Different crystal structures can have markedly different properties [4]. The products may transform to another polymorph over time or under different process conditions, which can reduce shelf life and severely disrupt production [5]. Researchers engaged in product development are thus anxious to determine all the possible polymorphs of their product as early as possible. A very good example of the importance of polymorphism is in the area of pigments and dyes. In the past several years there has been an increasing interest in the polymorph prediction of crystal structures on the basis of molecular information. Different methods have been developed to generate possible crystal structures [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. All researches agree that there will always be a large number of possible crystal structures, as judged from their lattice energy, and nearly all are successful in finding the experimental structure. The experimentally observed structure is generally very close in energy to the global minimum in the lattice energy [22].
In this study we investigate the ability of the method to predict correctly the possible polymorphs of L-ascorbic acid [23]. Now a systematic ab initio search is feasible. Our method uses a systematic search of parameters space, whereas many other published method rely on some sort of random search. We used the commercially available Biosym/Molecular Simulations Polymorph Predictor [7,8,9,24,25] to generate possible polymorphs for L-ascorbic acid.
2. Methods
The program [11], which performs brute-force grid search for possible crystal structures, has been completely revised will be referred to a Crystal Packer [7,8,9]. The code has been altered to handle triclinic, monoclinic, and orthorhombic space groups to be able to search the most abundant space groups for organic molecules. The crystallographic a-axis is chosen to coincide with the cartersian x-axis, and the b-axis lies in the x-y plane [26]. Then the crystallographic parameters to be varied are the orthogonal components of the cell axes (a, by and cz) and the components bx, cx and cy, which determine the cell angles. For a monoclinic structure, bx and cy are zero; for an orthorhombic structure, cx also is zero. The search is limited to densely packed structures by calculating each starting value of cz from an estimated cell volume, which is not very critical [26]. The component bx has to be varied from zero up to a length of a to find all unique setting of the a-b plane. Likewise, cx has to be varied from zero to a and cy from zero to by. Into each trial cell one independent rigid molecule is placed, with center-of mass coordinates x, y, z. The search proceeds by optimizing these starting parameters roughly, using a few simple steepest descent steps followed by some regular conjugate gradient minimization [26]. Equivalent structures must be removed from this list using a fast clustering algorithm [27]. Because these may differ in choice of unit cell, it is not always immediately obvious whether or not two structures are equivalent. This algorithm tests for equivalence between two structures by checking whether or not the rotation necessary to transform one orientation into the other corresponds to a cell transformation allowed by space-group symmetry [27]. This test allows for deviations of magnitude EN[5] from integer values in the transformation matrix elements. A final test compares all the components of the cell axes and the molecular centers of gravity after the transformation of one structure into the other, allowing for a tolerance. The second step of the procedure consists of a further rigid-body refinement. To reduce the computational effort it is worthwhile to perform an intermediate round of clustering when the structures are not yet properly minimized. This is followed by the final minimization, until a proper lattice-energy minimum is reached. The resulting lists of possible crystal structures are once more clustered. This is extremely important to limit the number of possible crystal structures, but clustering will be hindered by structures that have not yet completely converged to the same minimum. So, the final energy minimization must be continued until complete convergence is reached, for which it is necessary to use double precision and a very sensitive convergence criterion (0.001 kJ/ A or less for the rms gradient of energy). As a cut-off distance for summing the intermolecular interaction is used, is fixed pair list is necessary to avoid discontinuities in the energy between successive minimization steps. This pair list is updated only when a parameters has changed more than a certain threshold. Structures that are found after the final minimization are minima in the energy surface under the constraints of the imposed space-group symmetry. They are not necessarily minima in the absence of those constraints; that is, the crystal does not have to be at minimum energy with respect to the extra degrees of freedom. When the space-group constraints are released by expanding the structures to P1 all the force on the molecules 6 remain zero by virtue of symmetry, including the force field in the direction of the extra degrees of freedom. The structures are minima or saddle points in the complete crystal-energy surface. Whether a structure is a true minimum can be tested by minimizing the crystal structures after an expansion to P1 [27].
After minimizing, the structures must be analyzed for the remaining symmetry using a program like PLATON [27]. Clustering of these structures, which may have several independent molecules (Z > 1), cannot be performed with the clustering algorithm [28]. An atom-atom distance based clustering strategy has been adopted for these cases. For each molecule in the P1 unit cell a list of atom-atom distances within a certain distance is made. First, within one crystal structure the lists for all the separate molecules to be considered are symmetry related. This gives the number of independent molecules in the unit cell, albeit based on local rather than crystallographic symmetry. Second, different structures which have been assigned the same number of independent molecules are compared, again allowing for a maximum difference in the distances. This code is considerably slower than the clustering algorithm [27] , but this is not a problem as the number of structures to be considered at this stage is not excessive. It is conceivable that two energy minima are separate by a fairly low barrier. In such a case the least favorable one can easily convert into the other, thus reducing the number of possible crystal structures [28]. To search this possibility we subjected the structures to a primitive molecular dynamics simulation, where the space-group symmetry is enforced at every time step [28]. This is, of course, very unrealistic, but any other approach would make excessive demands on computer time. These simulations are just meant to “shake up” the 7 structures to overcome barries of the order of kT. Simulations were carried out at constant temperature (200 K, but starting from 10K) and pressure (1 atm), using the weak-coupling method [28].
A crystal-packing prediction with the Polymorph Predictor consists of a sequence of several steps that can be carried out automatically. It produces a set of predicted crystal structures for a single space group with a given number of independent molecules in the unit cell, and a given starting molecular geometry. The program can handle all 230 space groups, with any number of independent molecules in the cell [28].
In the first stage of the prediction, a Monte Carlo / simulated annealing packing algorithm generates starting structures, treating the molecule as a rigid unit. In each Monte Carlo step, all relevant parameters are changed randomly. To suppress crystal evaporation the parameters defining the spatial extension of the crystal are handled. The Monte Carlo simulation takes 4000-5000 steps, resulting in about 2000 accepted structures. The simulated annealing producer is applied to improve the efficiency of finding the most promising starting structures. During the Monte Carlo simulation, the temperature parameter is first increased after each step, until a specified number of consecutive trials have been accepted. This will take 50-100 steps, and the temperature parameter will rise to about 10,000 K during the rest of the Monte Carlo simulation, the temperature is slowly decreased until it reaches a certain minimum value (usually 200 K). The second stage of the prediction consists of clustering of the structures produced by the Monte Carlo run. Clustering is based on interatomic distance, discriminating by force field type. For each structure a list of interatomic distances within a certain cut off is 8 made for each combination of atom types. The number of clusters formed in this case is around 1000. The third step of the calculations consists of a full geometry minimization of one structure from each cluster, optimizing atomic atomic coordinates and cell parameters, but imposing space-group symmetry. Finally, a clustering is performed on all minimized structures, identical to the clustering just described, leaving somewhere between 10 and 300 different structures, within an energy range of about 50 kJ/mol.
3. Force Fields
General force constants and geometry parameters for the Dreiding force field are based on simple hybridization rules rather than on specific combinations of atoms. The Dreiding force field is a purely diagonal force field with harmonic valence terms and a Cosine-Fourier expansion torsion term. The Van der waals interaction are described by the Lennard-Jones potential. Electrostatic interactions are described by atomic monopoles and a screened (distance-dependent) coulombic. In the polymorph predictor runs, the DREIDING force field [29], as implemented in Cerius2 ( Cerius2 is a trademark of Biosym/Molecular Simulation), was used. Calculations were carried out using several types of atomic charges like MNDO ESP charges, as suggested by Besler et al. 30, as well as ESP charges derived from ab initio calculation using an STO-3G basis set, scaled by an empirical factor. Hydrogen bonding is described by an explicit Lennard-Jones 12-10 potential. The Dreiding force field [9,29,31] has good coverage for organic, biological and main-group inorganic molecules, and it is a good, robust, all-purpose force field. It can be used for structure prediction and dynamics calculations on organic, biological, and main group inorganic molecules.
It is an excellent general-purpose force field. It is based on element, hybridization, and connectivity. The universal force field was parameterized for the full periodic table and has been validated for main group compounds.
4. Result and Discussion
The investigation for possible crystal structures [32] was carried out in the six most abundant space groups, up to the space group of the experimental structure (P21). Together, they cover 85% of the crystal structures in the Cambridge Crystallographic Database. The DREIDING force field was used and the molecular geometry was taken to be the minimized structure of the free L-ascorbic acid molecule in that force field. In the grid search the cell parameters and center-mass coordinates were varied in steps 1 Å, and the angles in steps of 30°. The numbers of resulting structures are given as No in Table I. The clustering procedure reduced those numbers to N1. The tolerances here were εN = 0.2 and εA = 1 Å [27]. Throughout the rest of the procedure clustering tolerances of .N = 0.3 and .A = 0.3 Å were used 27. Continuing the energy minimization with the DREIDING force field [31] the cut-off radius extended to 10 Å. The number of resulting structures after clustering are given as N2 in Table I. The final rigid-body minimization to a proper energy minimum was done using a 10 cut-off radius of 12 Å. The numbers of structures that remained after clustering are reported as N3 in Table I. It can be seen that continuing the minimization of the approximately minimized structures (N2) until complete convergence had been reached (N3) proved very valuable. That is, after a full geometry relaxation and clustering, the numbers N4 in Table I(. R) remained. Inspection of some structures that are unstable in a flexible minimization revealed that they have the L-ascorbic acid molecule located on a crystallographic mirror plane. In a rigid-body approximation there apparently existed a energy barrier for rotating the molecule out of the this plane, which has absent in the case of a flexible molecule. Indeed, in the first case, a moderate distortion of the symmetry was seen to return to the starting position, showing it to be a true minimum; in the second case, even a very small distortion resulted in a reorientation of the molecule, showing that the structure now represented a saddle point. On visual inspection it was far from obvious how the flexibility was used to removed such an energy barrier. The grid-search algorithm (was) remove has succeeded in finding the minimum corresponding to the experimental structure and, as result, the experimental structure was present in the final structure lists of all force fields. However, the structural parameters for the energy-minimized experimental structures were different for the different force fields. The cell axes are given in Table II, together with the ranking of the experimental structure. It is seen that all potentials resulted in a reasonably low ranking with an energy difference that is of the same order of magnitude as what one could reasonably expect for the uncertainties of the potentials [11. Predictions of crystal structure were carried out in the same six space groups. The DREIDING force field, as described previously, was used. So the search strategy as well as the force field were different, allowing a comparison with the other force field procedure on both levels. The isolated molecule was optimized at the STO-3G level using GAUSSIAN-94 [30]. A number of standard settings for all simulation steps in the Polymorph Predictor is available. The Monte Carlo search level was set to find, and the clustering search level was also set to find, the tolerance was set to 0.1, and the limit on the number of generated clusters was removed. All structures were fully minimized, taking the rms force of 0.001 kcal/ Å as convergence criterion. Van der Waals and Coulomb interactions were calculated using the Ewald summation method. The numbers of structures that remained after minimization and the final round of clustering are given as in Table I.
The experimental structure was found, but the predicted unit cell was significantly larger. Axes differed by up to almost 20% (Table II). Another striking difference was the hydrogen bond distance that was too large (Table II), which would explain why the structures were too weakly bound (Table III). The energy of the experimental structure was again high. The larger number of low-energy structures was found using this force field than when using the others.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table I.
The results of search of crystal structures [32] of L-ascorbic acid
| P1 | P-1 | P21 | P2/c | C2 | P21212 | |
| No | 4066 | 4221 | 7000 | 4459 | 6469 | 5889 |
| N1 | 500 | 2372 | 879 | 2430 | 500 | 3564 |
| N2 | 533 | 500 | 453 | 1872 | 250 | 1753 |
| N3 | 15 | 10 | 30 | 50 | 26 | 14 |
| N4 | 8 | 11 | 18 | 15 | 7 | 37 |
Table II.
Cell Axes, Hydrogen-Bond Geometries, and Lattice-Energy Minima of L-Ascorbic Acid Corresponding to the Experimental Structure, for Different Force Fields [31].
| a (Å) | b (Å) | c(Å) | O---O (Å) | O____H------ O (°) | ΔE | |
| Exp. | 6.12 | 3.61 | 11.88 | 2.75 | 160 | - |
| DREIDING | 5.87 | 4.79 | 10.25 | 2.078 | 175 | 1.3 |
| AMBER | 6.05 | 3.45 | 11.07 | 2.893 | 153 | 1.8 |
| UNIVERSAL | 5.97 | 3.56 | 11.30 | 2.866 | 155 | 2.1 |
| COMPASS | 6.08 | 3.59 | 11.68 | 2.082 | 171 | 1.7 |
Table III.
Lattice Energy and Number of Low-Energy structures of vitamin C ( L-Ascorbic Acid) molecule.
| P1 | P-1 | P21 | P2/c | C2 | P21212 | |
| DREIDING | -35.8 | -34.8 | -33.4 | -29.4 | -34.3 | -28.7 |
| N | 4 | 7 | 15 | 11 | 3 | 17 |
| AMBER | -64.3 | -63.1 | -64.9 | -62.3 | -61.8 | -63.7 |
| N | 5 | 6 | 3 | 17 | 23 | 4 |
| COMPASS | -43.5 | -42.7 | -43.1 | -41.8 | -43.4 | 42.9 |
| N | 2 | 3 | 12 | 4 | 22 | 3 |
| UNIVERSAL | -25.4 | -25.7 | -24.4 | -24.7 | -25.3 | 25.1 |
| N | 2 | 7 | 4 | 3 | 10 | 13 |
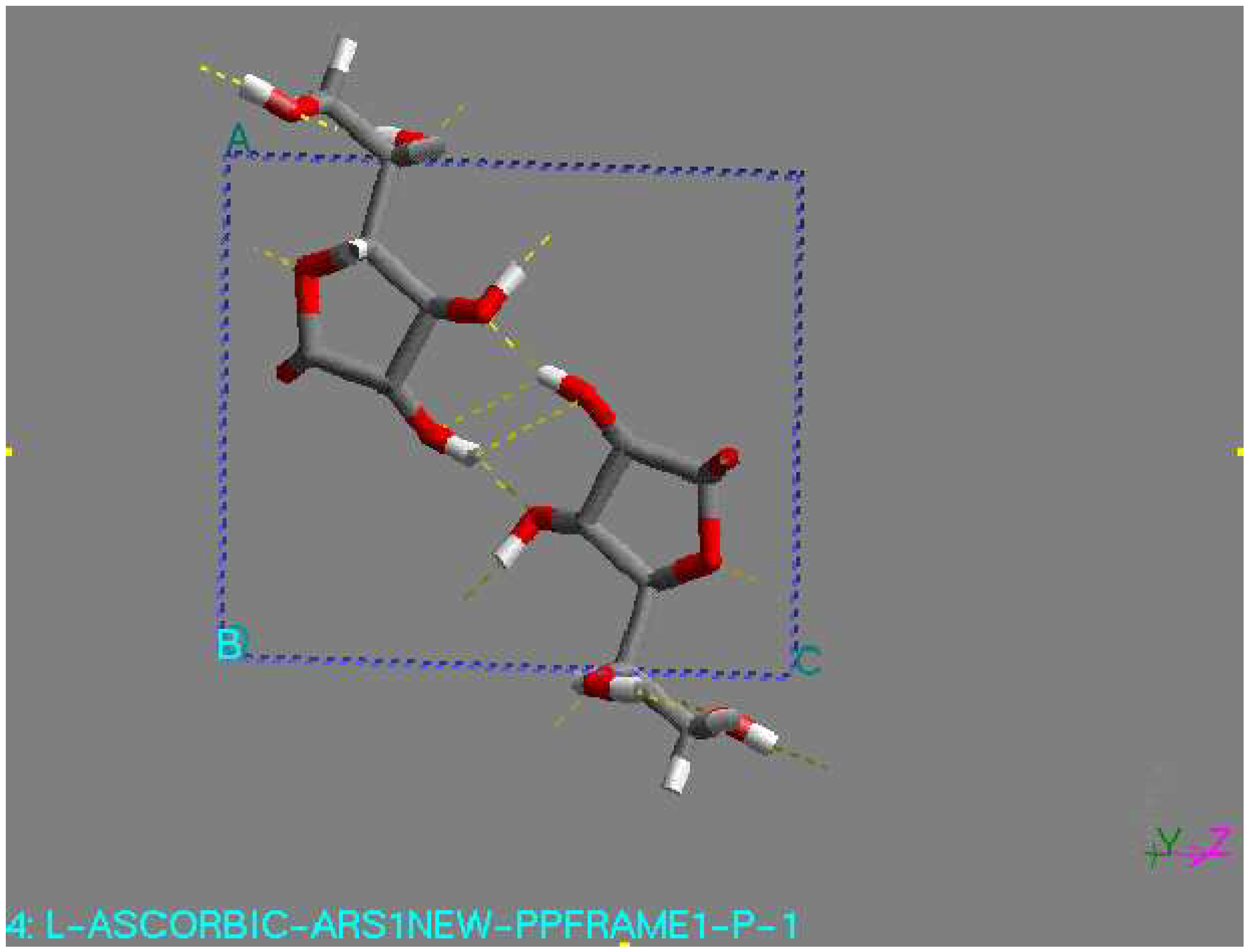
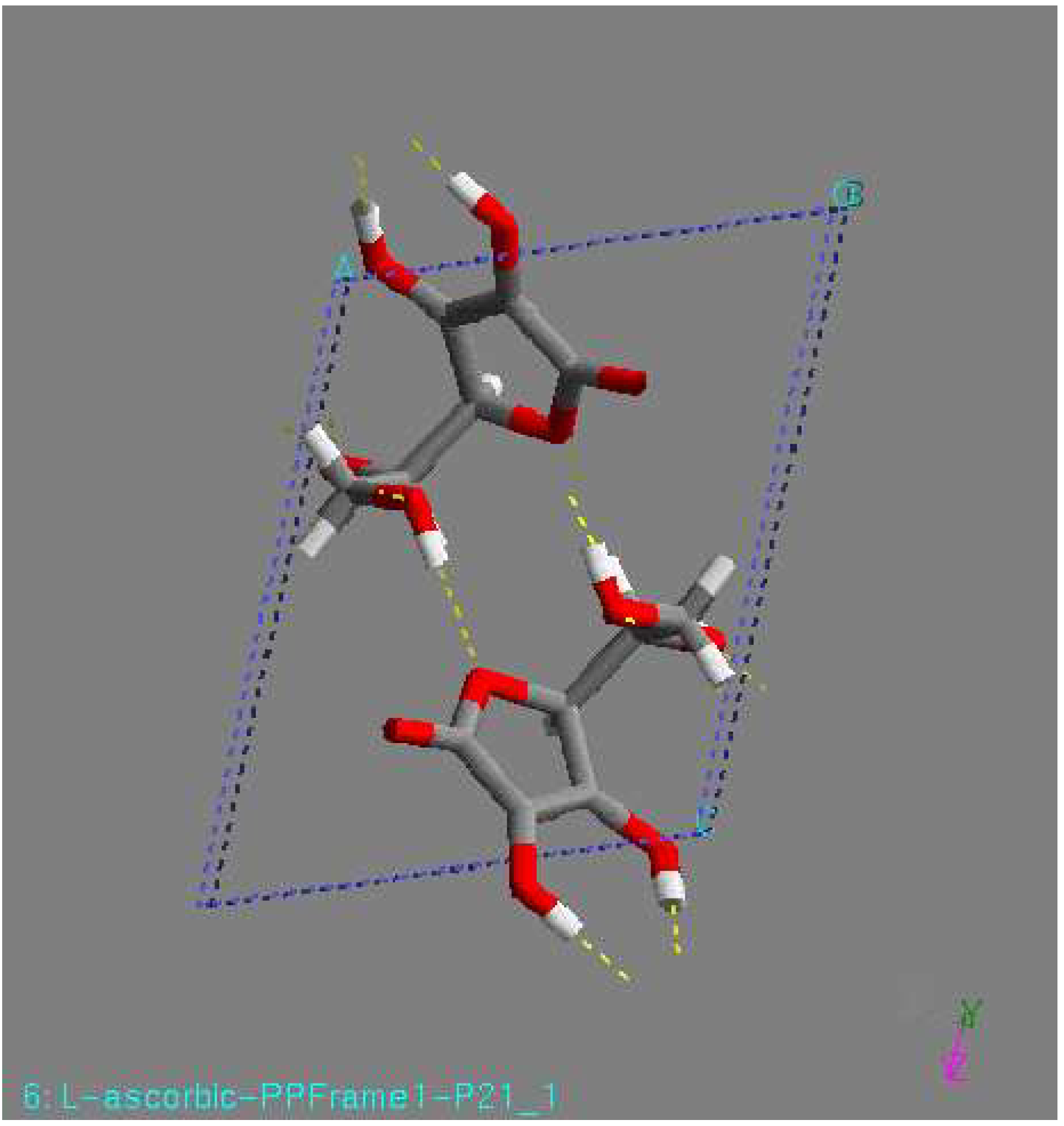
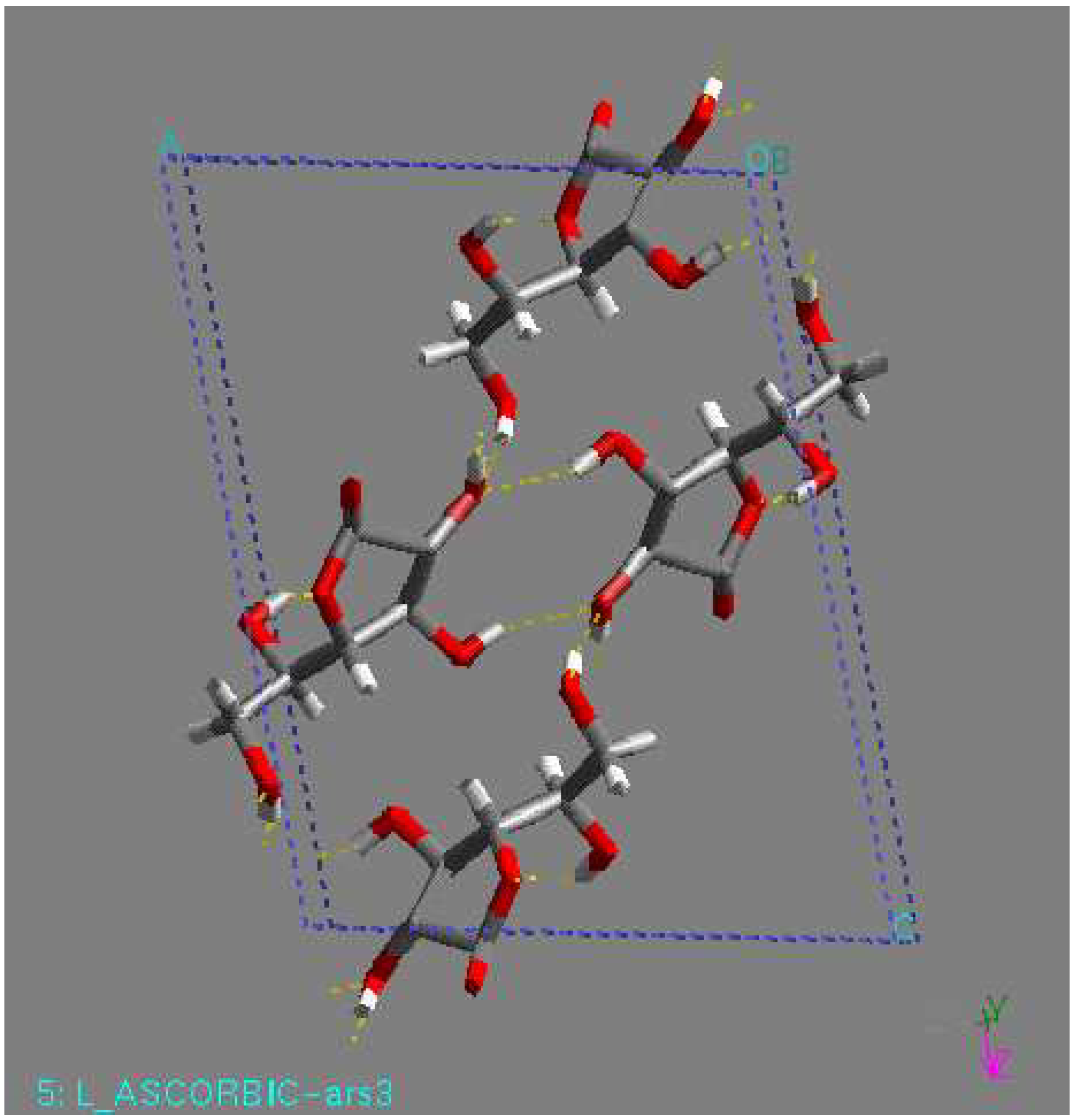
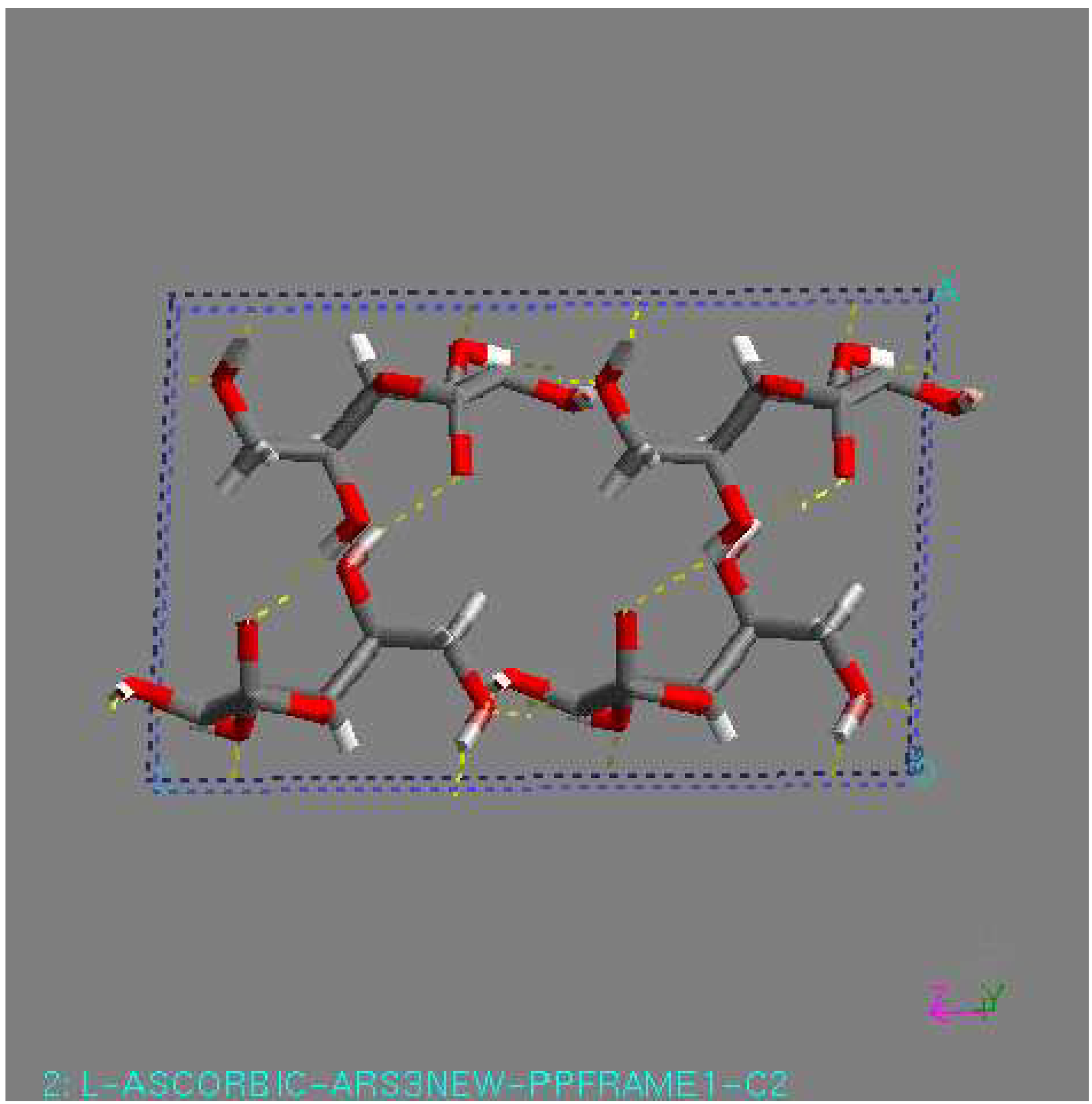
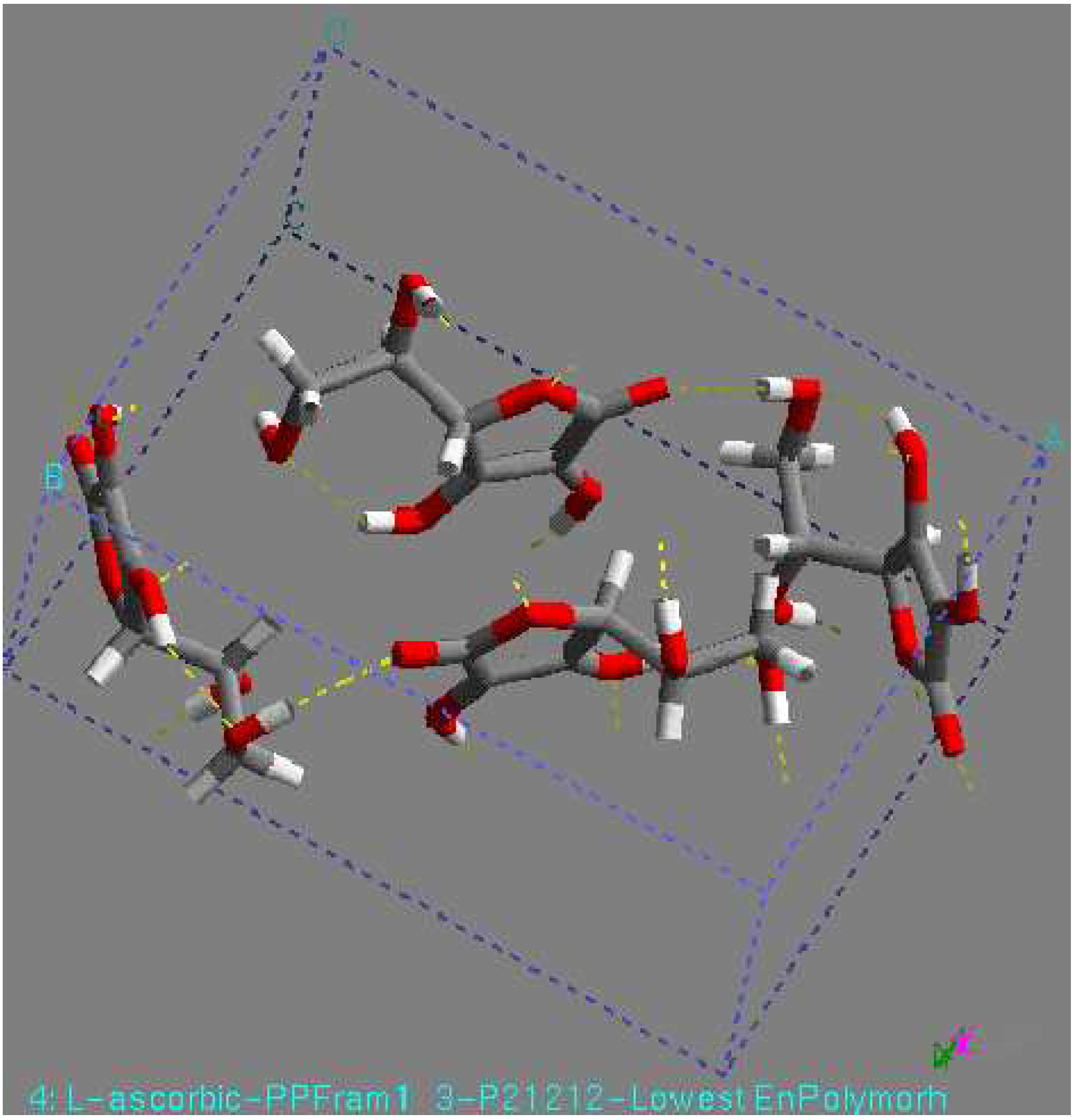
As an L-ascorbic acid molecule has internal symmetry, a crystal structure can have symmetry during minimization. The final crystal structure can be described in another space group with the molecule on a special position. Indeed, this happened in a number of cases. An example is the structure with the lowest lattice energy in AMBER [31], which was also found among the relatively stable structures in the other force fields. This is a P1 structure, with the L-ascorbic acid molecule (see Fig. 1). As an indication of the search procedure it is worth mentioning that this structure was found in all space groups considered that are crystallographic subgroups. Equivalent structures of higher symmetry are not automatically clustered when they are found in different space groups. Within one space-group list it may also happen that two settings of such a structure are not clustered; that is , the transformation needed for the clustering may be forbidden in assumed space group of lower symmetry. All structures within 15 kJ/mol (higher in energy than) of the global minimum in the COMPASS force field [31] were expanded to P1 unit cells and subsequently minimized using a cut-off radius of 12 Å. As no rigid-body minimization was implemented for more than one in dependent molecule, this was carried out with full geometry relaxation. During such a minimization the structures can lose some of their symmetry. Some space groups, like P21212, P21 maintained their original symmetry in all cases. However, especially in P2/c and C2, a large number of structures lost some of their symmetry. Those structures ended up in other space groups having two or more independent molecules. However, quite a large number of structures resulted again having one independent molecule in a different space group, like P1 (see Fig. 1). For example, the structure with the lowest 14 energy to come from the list of space group P1 had one independent molecule, with the space group changed to P-1 had two molecule (see Fig 2), with the space group P2/c had four molecule (see Fig. 3), with space group P21 had two molecule (see Fig. 4), and with space group P21212 had four molecules in unit cell (see Fig. 5).
Figure 1.
P-1 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis
Figure 1.
P-1 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis

Figure 2.
P21 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.
Figure 2.
P21 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.

Figure 3.
P2/c structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.
Figure 3.
P2/c structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.

Figure 4.
C2 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.
Figure 4.
C2 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.

Figure 5.
P21212 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.
Figure 5.
P21212 structure, found by minimization. This structure had the lowest lattice energy in the DREIDING force field. View along the c-axis.

A different question to consider all the structures with Z > 1, as their energy can be rather low. Perhaps the best thing to do is to recall the statistics of the Cambridge Structural Database, which show us that only 10% of all crystal structures [32] have more than one independent molecule. Based on this percentage one can more easily concentrate on the Z = 1 structures.
5. Conclusion
Researchers engaged in product development are thus anxious to determine all the possible polymorphs of their product as early as possible . The past several years there has been an increasing interest in the polymorph prediction of crystal structures on the basis of molecular information [32]. Different methods have been developed to generate possible crystal structures. All researches agree that there will always be a large number of possible crystal structures, as judged from their lattice energy, and nearly all are successful in finding the experimental structure. The experimentally observed structure is generally very close in energy to the global minimum in the lattice energy. It appears likely that, for many molecules, there is a range of energetically possible structures, and that the many other factors that affect the kinetics of crystal growth are responsible for selecting those that can be found experimentally. It would be necessary to look at such factors before anyone can expects to reach to a correct prediction.
Acknowledgements
This project was supported by Kafkas University, Kars, Turkey, TR and Highly Filled Materials Institute (HFMI), Stevens Institute of Technology, USA.
References
- Grimbergen, R. F.P.; Reediijk, M.F.; Meekes, H.; Bennema, P. J. Phys. Chem. B. 1998, 102, 2646.
- Meekes, H.; Bennema, P.; Grimbergen, R. F. P. Acta Cryst. B. 1998, 54, 501.
- Grimbergen, R. F. P.; Bennema, P.; Meekes, H. Acta Cryst. A. 1999, 55, 84.
- Verwer, P.; Leusen, F. J. J. Reviews in Comput. Chem. 1998, 12, 327.
- Myerson, A.; Jang, S. J. Cryst. Growth. 1995, 156, 459.
- Gavezzotti, A. J. Am. Chem. Soc. 1991, 133, 4622.
- Karfunkel, H.R.; Gdanitz, R. J. J. Comput. Chem. 1992, 13, 1171.
- Karfunkel, H. R.; Leusen, F. J. J. Speedup. 1993, 6, 43.
- Karfunkel, H. R.; Leusen, F. J. J.; Gdanitz, R. J. J. Comput.-Aid. Mat. Des. 1993, 1, 177.
- Holden, J. R.; Du, Z.; Ammon, H. L. J. Comput. Chem. 1993, 14, 422.
- Van Eijck, B. P.; Mooij, W. T. M.; Kroon, J. Acta Cryst. B. 1995, 51, 99.
- Shoda, T.; Yamahara, K.; Okazaki, K.; Williams, D. E. J. mol. Struct. (TheoChem). 1995, 333, 267.
- Shoda, T.; Williams, D. E. J. mol. Struct. (TheoChem). 1995, 357, 1.
- Gibson, K. D.; Scheraga, H. A. J. Phys. Chem. 1995, 99, 3765.
- Tajima, N.; Tanaka, T.; Arikawa, T.; Sakurai, T.; Teramae, S.; Hirano, T. Bull. Chem. Soc. Jpn. 1995, 68, 519.
- Arikawa, T.; Tajima, N.; Tsuzuki, S.; Tanabe, T.; Hirano, T. J. Mol. Struct. (TheoChem) 1995, 339, 115.
- Gavezzotti, A. Acta Cryst. B. 1996, 52, 201.
- Schmidt, M. U.; Englert, U. J. Chem. Soc. (Dalton Trans.). 1996, 2077.
- Chaka, A. M.; Zaniewski, R.; Youngs, W.; Tessier, C.; Klopman, G. Acta Cryst. B. 1996, 52, 165.
- Williams, D. E. Acta Cryst. A. 1996, 52, 326.
- Wavak, R. J.; Gibson, K. D.; Liwo, A.; Scheraga, H. A. Proc. Natl. Acad. Sci. 1996, 93, 1743.
- Wavak, A. V.; Pivina, T. S.; Arnautova, E. A. J. Mol. Struct. 1996, 378, 67.
- Jonsson, P. G. Acta Cryst. B. 1971, 27, 893.
- Gdanitz, R. J. Chem. Phys. Lett. 1992, 190, 391.
- Karfunkel, H. R.; Wu, Z. J.; Burkhard, A.; Rihs, G.; Sinnreich, D.; Buerger, H. M.; Stanek, J. Acta Cryst. B. 1996, 52, 555.
- Leusen, F. J. J. J. Cryst. Growth. 1996, 166, 900.
- Press, W. H.; Flannery, B. P.; Teukolsky, S. A.; Vetterling, W. T. Numerical Recipes, the Art of Scientific Computing; Cambridge University Press: Cambridge, UK, 1989; p. 301. [Google Scholar]
- VanEijck, B. P.; Kroon, J. J. Comput. Chem. 1997, 18, 1036.
- Spek, A. L. Acta Cryst. A. 1990, 46, 34.
- Rappe, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A.; Skiff, W. M. J. Am. Chem. Soc. 1992, 114, 10024.
- McQuaid, M.J.; Sun, H.; Rigby, D. J. Comput. Chem. 2004, 25, 61.
- Panagiotis, G.; Karamertzanis; Constantinos, C.; Pantelides. J. Comput. Chem. 2005, 36, 304.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Arslantas, A.; Ermler, W.C.; Yazici, R.; Kalyon, D.M. Study of Polymorph Prediction For L-Ascorbic Acid. Int. J. Mol. Sci. 2005, 6, 291-302. https://doi.org/10.3390/i6120291
AMA Style
Arslantas A, Ermler WC, Yazici R, Kalyon DM. Study of Polymorph Prediction For L-Ascorbic Acid. International Journal of Molecular Sciences. 2005; 6(12):291-302. https://doi.org/10.3390/i6120291
Chicago/Turabian StyleArslantas, Ali, Walter C. Ermler, Rahmi Yazici, and Dilhan M. Kalyon. 2005. "Study of Polymorph Prediction For L-Ascorbic Acid" International Journal of Molecular Sciences 6, no. 12: 291-302. https://doi.org/10.3390/i6120291