Synthesis and GIAO NMR Calculations for Some Novel 4-Heteroarylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives: Comparison of Theoretical and Experimental 1Hand 13C- Chemical Shifts

Abstract
:Introduction

Experimental
General
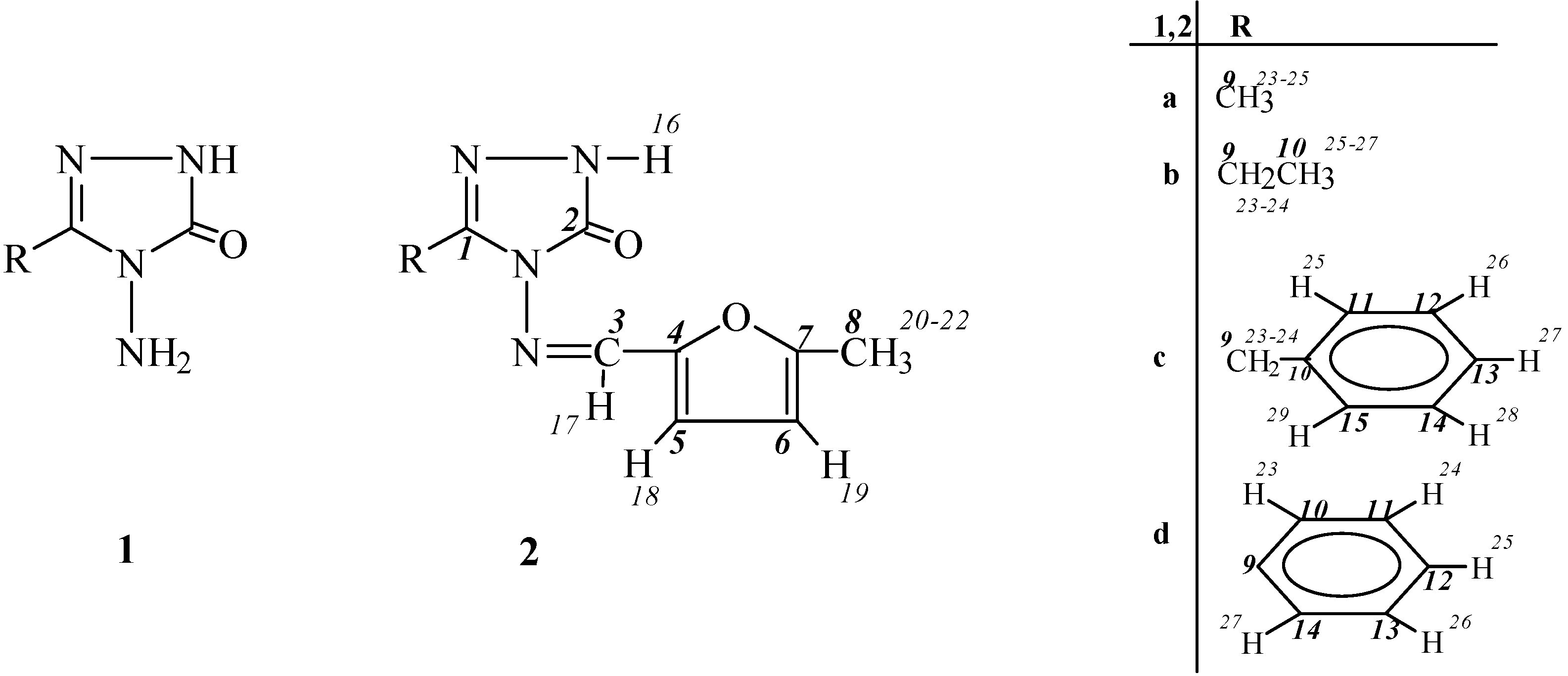
General Method for the Preparation of 3-Alkyl(aryl)-4-(5-methyl-2-furylmethylenamino)-4,5-dihydro-1H-1,2,4-triazol-5-ones (2)
3-Methyl-4-(5-methyl-2-furylmethylenamino)-4,5-dihydro-1H-1,2,4-triazol-5-one (2a).
3-Ethyl-4-(5-methyl-2-furylmethylenamino)-4,5-dihydro-1H-1,2,4-triazol-5-one (2b).
3-Benzyl-4-(5-methyl-2-furylmethylenamino)-4,5-dihydro-1H-1,2,4-triazol-5-one (2c).
3-Phenyl-4-(5-methyl-2-furylmethylenamino)-4,5-dihydro-1H-1,2,4-triazol-5-one (2d).
Computational Methods

{kind=link}
{kind=link}
{kind=link}
| Nuclei | Experimental | B3LYP/6-311G | Diff | HF/6-311G | Diff |
|---|---|---|---|---|---|
| C-2 | 153.10 | 156.22 | -3.12 | 166.13 | -13.03 |
| C-1 | 145.90 | 157.21 | -11.31 | 163.06 | -17.16 |
| C-9 | 13.02 | 14.51 | -1.49 | 14.39 | -1.37 |
| C-3 | 148.64 | 153.12 | -4.48 | 168.01 | -19.37 |
| C-5 | 121.35 | 131.09 | -9.74 | 135.58 | -14.23 |
| C-4 | 144.78 | 161.44 | -16.66 | 153.37 | -8.59 |
| C-7 | 158.10 | 169.97 | -11.87 | 170.68 | -12.58 |
| C-6 | 110.90 | 118.13 | -7.23 | 115.65 | -4.75 |
| C-8 | 15.38 | 15.70 | -0.32 | 15.74 | -0.36 |
| H-16 | 11.81 | 6.98 | 4.83 | 6.96 | 4.85 |
| H-23 | 2.23 | 2.10 | 0.13 | 2.30 | -0.07 |
| H-24 | 2.23 | 2.07 | 0.16 | 2.04 | 0.19 |
| H-25 | 2.23 | 2.55 | -0.32 | 2.60 | -0.37 |
| H-17 | 9.50 | 8.50 | 1.00 | 9.32 | 0.18 |
| H-18 | 7.06 | 6.65 | 0.41 | 7.25 | -0.19 |
| H-19 | 6.34 | 6.21 | 0.13 | 6.37 | -0.03 |
| H-20 | 2.38 | 2.59 | -0.21 | 2.67 | -0.29 |
| H-21 | 2.38 | 2.53 | -0.15 | 2.59 | -0.21 |
| H-22 | 2.38 | 2.19 | 0.19 | 2.32 |
| Nuclei | Experimental | B3LYP/6-311G | Diff | HF/6-311G | Diff |
|---|---|---|---|---|---|
| C-2 | 153.25 | 156.43 | -3.18 | 166.21 | -12.96 |
| C-1 | 148.71 | 161.11 | -12.40 | 166.41 | -17.70 |
| C-9 | 20.26 | 24.85 | -4.59 | 20.72 | -0.46 |
| C-3 | 149.65 | 153.17 | -3.52 | 168.08 | -18.43 |
| C-5 | 121.09 | 131.05 | -9.96 | 135.42 | -14.33 |
| C-4 | 144.77 | 161.39 | -16.62 | 153.45 | -8.68 |
| C-7 | 158.03 | 169.85 | -11.82 | 170.59 | -12.56 |
| C-6 | 110.85 | 117.91 | -7.06 | 115.63 | -4.78 |
| C-8 | 15.33 | 15.70 | -0.37 | 15.74 | -0.41 |
| C10 | 11.62 | 10.00 | 1.62 | 9.64 | 1.98 |
| H-16 | 11.82 | 6.99 | 4.83 | 6.95 | 4.87 |
| H-23 | 2.62 | 2.43 | 0.19 | 2.22 | 0.40 |
| H-24 | 2.62 | 2.99 | -0.37 | 2.84 | -0.22 |
| H-17 | 9.49 | 8.48 | 1.01 | 9.32 | 0.17 |
| H-18 | 7.03 | 6.66 | 0.37 | 7.22 | -0.19 |
| H-19 | 6.32 | 6.21 | 0.11 | 6.37 | -0.05 |
| H-20 | 2.37 | 2.59 | -0.22 | 2.67 | -0.30 |
| H-21 | 2.37 | 2.54 | -0.17 | 2.58 | -0.21 |
| H-22 | 2.37 | 2.20 | 0.17 | 2.31 | 0.06 |
| H-25 | 1.18 | 1.02 | 0.16 | 1.09 | 0.09 |
| H-26 | 1.18 | 1.45 | -0.27 | 1.60 | -0.42 |
| H-27 | 1.18 | 1.50 | -0.32 | 1.55 | -0.37 |
| Nuclei | Experimental | B3LYP/6-311G | Diff | HF/6-311G | Diff |
|---|---|---|---|---|---|
| C-2 | 153.09 | 159.30 | -6.21 | 166.94 | -13.85 |
| C-1 | 147.86 | 159.77 | -11.91 | 164.70 | -16.84 |
| C-9 | 32.73 | 37.84 | -5.11 | 32.66 | 0.07 |
| C-3 | 148.73 | 148.40 | 0.33 | 157.99 | -9.26 |
| C-5 | 120.96 | 118.92 | 2.04 | 122.18 | -1.22 |
| C-4 | 144.72 | 164.44 | -19.72 | 158.32 | -13.60 |
| C-7 | 158.11 | 170.31 | -12.20 | 169.14 | -11.03 |
| C-6 | 110.92 | 117.05 | -6.13 | 115.29 | -4.37 |
| C-8 | 15.37 | 15.62 | -0.25 | 15.69 | -0.32 |
| C-10 | 137.41 | 142.28 | -4.87 | 144.01 | -6.60 |
| C-11 | 130.77 | 137.36 | -6.59 | 140.57 | -9.80 |
| C-12 | 130.14 | 134.64 | -4.50 | 138.41 | -8.27 |
| C-16 | 128.47 | 133.57 | -5.10 | 137.07 | -8.60 |
| C-14 | 130.14 | 134.64 | -4.50 | 138.41 | -8.27 |
| C-15 | 130.77 | 137.36 | -6.59 | 140.57 | -9.80 |
| H-16 | 11.97 | 6.94 | 5.03 | 6.92 | 5.05 |
| H-23 | 4.00 | 3.91 | 0.09 | 3.93 | 0.07 |
| H-24 | 4.00 | 3.91 | 0.09 | 3.93 | 0.07 |
| H-17 | 9.49 | 10.50 | -1.01 | 10.73 | -1.24 |
| H-18 | 7.05 | 7.15 | -0.10 | 7.56 | -0.51 |
| H-19 | 6.33 | 6.20 | 0.13 | 6.38 | -0.05 |
| H-20 | 2.38 | 2.58 | -0.20 | 2.65 | -0.27 |
| H-21 | 2.38 | 2.58 | -0.20 | 2.65 | -0.27 |
| H-22 | 2.38 | 2.15 | 0.23 | 2.27 | 0.11 |
| H-25 | 7.23 | 7.23 | 0 | 7.74 | -0.51 |
| H-26 | 7.33 | 7.33 | 0 | 7.84 | -0.51 |
| H-27 | 7.30 | 7.27 | 0.03 | 7.77 | -0.47 |
| H-28 | 7.33 | 7.33 | 0 | 7.84 | -0.51 |
| H-29 | 7.23 | 7.23 | 0 | 7.74 | -0.51 |
| Nuclei | Experimental | B3LYP/6-311G | Diff | HF/6-311G | Diff |
|---|---|---|---|---|---|
| C-2 | 153.26 | 159.80 | -6.54 | 167.27 | -14.01 |
| C-1 | 147.63 | 156.02 | -8.39 | 162.39 | -14.76 |
| C-9 | 130.26 | 134.41 | -4.15 | 134.60 | -4.34 |
| C-3 | 148.59 | 148.96 | -0.37 | 158.14 | -9.55 |
| C-5 | 121.64 | 119.29 | 2.35 | 122.35 | -0.71 |
| C-4 | 146.26 | 164.62 | -18.36 | 158.57 | -12.31 |
| C-7 | 158.44 | 170.46 | -12.02 | 169.08 | -10.64 |
| C-6 | 111.06 | 117.09 | -6.03 | 115.29 | -4.23 |
| C-8 | 15.39 | 15.73 | -0.34 | 15.69 | -0.30 |
| C-10 | 129.64 | 134.89 | -5.25 | 141.15 | -11.51 |
| C-11 | 129.64 | 134.20 | -4.56 | 136.67 | -7.03 |
| C-12 | 131.82 | 136.36 | -4.54 | 141.72 | -9.90 |
| C-13 | 128.52 | 133.61 | -5.09 | 135.51 | -6.99 |
| C-14 | 130.26 | 135.89 | -5.63 | 142.08 | -11.82 |
| H-16 | 12.37 | 7.27 | 5.10 | 7.27 | 5.10 |
| H-17 | 9.39 | 10.72 | -1.33 | 10.87 | -1.48 |
| H-18 | 7.12 | 7.16 | -0.04 | 7.48 | -0.36 |
| H-19 | 6.36 | 6.22 | 0.14 | 6.36 | 0 |
| H-20 | 2.36 | 2.59 | -0.23 | 2.65 | -0.29 |
| H-21 | 2.36 | 2.59 | -0.23 | 2.65 | -0.29 |
| H-22 | 2.36 | 2.16 | 0.20 | 2.26 | 0.10 |
| H-23 | 7.90 | 8.28 | -0.38 | 8.70 | -0.80 |
| H-24 | 7.54 | 7.42 | 0.12 | 7.92 | -0.38 |
| H-25 | 7.50 | 7.39 | 0.11 | 8.04 | -0.54 |
| H-26 | 7.54 | 7.41 | 0.13 | 7.90 | -0.36 |
| H-27 | 7.92 | 8.43 | -0.51 | 8.57 | -0.65 |
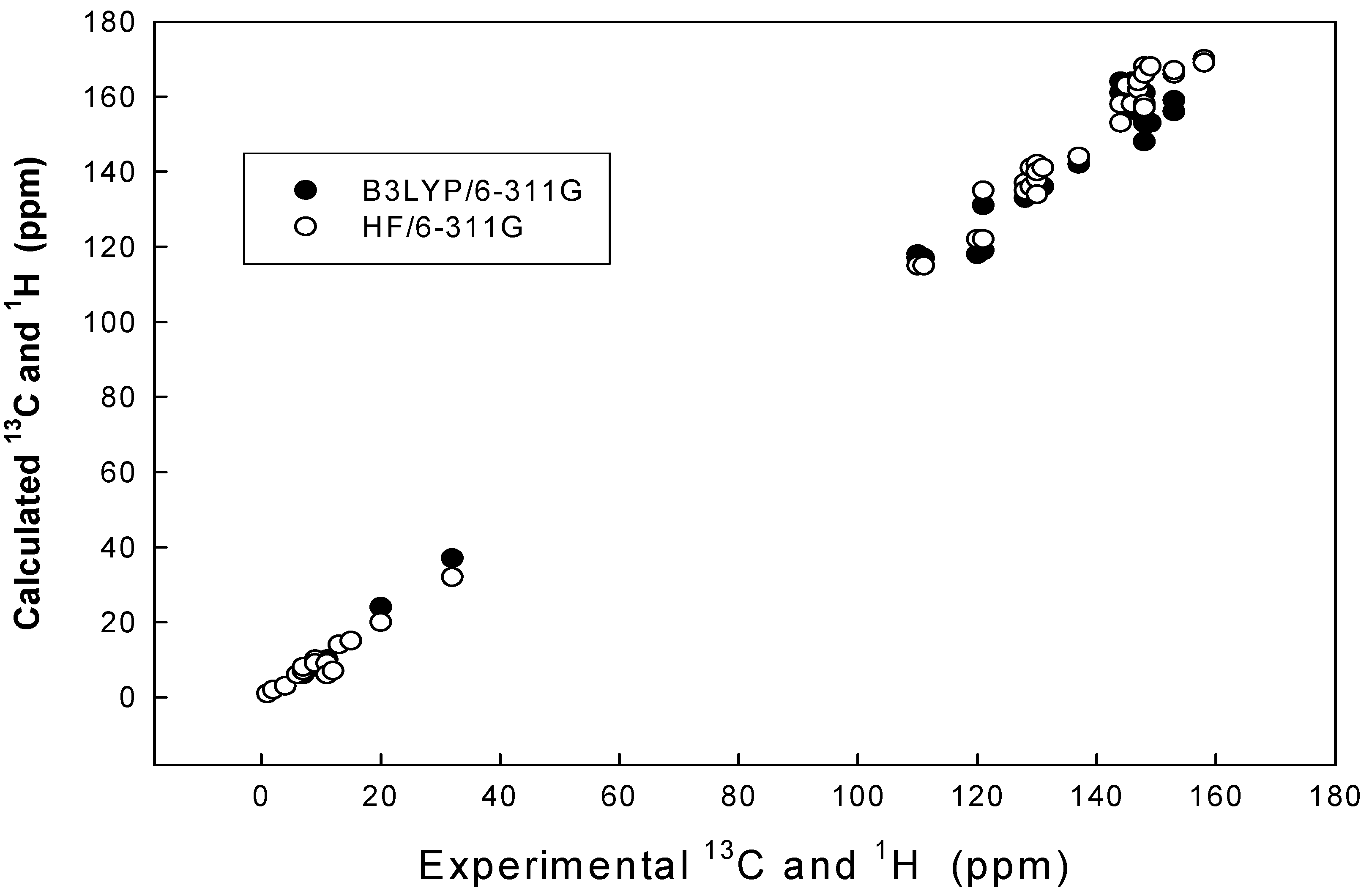
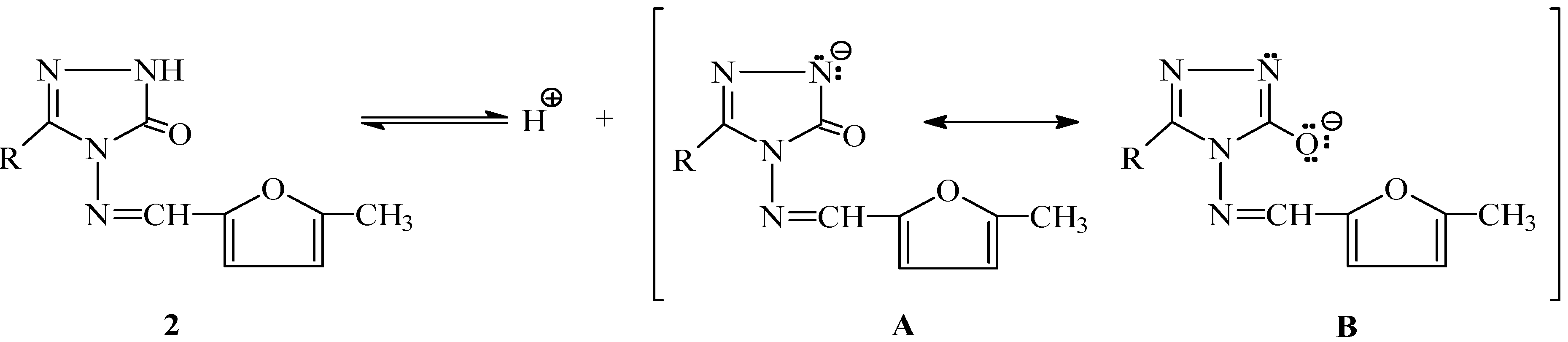
Results and Discussion

| Compd. no | Parameter | B3LYP/6-311G 13C | B3LYP /6-311G 1H | HF/6-311G 13C | HF/6-311G 1H |
|---|---|---|---|---|---|
| 2a | a | 0.386 | 1.049 | -0.879 | 1.087 |
| b | 1.062 | 0.656 | 1.098 | 0.691 | |
| r | 0.998 | 0.934 | 0.998 | 0.912 | |
| SE | 4.408 | 0.954 | 4.012 | 1.179 | |
| 2b | a | 0.313 | 0.847 | -2.282 | 0.875 |
| b | 1.062 | 0.678 | 1.107 | 0.716 | |
| r | 0.998 | 0.944 | 0.998 | 0.926 | |
| SE | 4.572 | 0.887 | 3.595 | 1.090 | |
| 2c | a | 0.124 | 1.250 | -3.353 | 1.345 |
| b | 1.049 | 0.750 | 1.094 | 0.777 | |
| r | 0.993 | 0.870 | 0.997 | 0.853 | |
| SE | 5.180 | 1.251 | 3.166 | 1.400 | |
| 2d | a | -2.657 | 1.280 | -3.340 | 1.345 |
| b | 1.065 | 0.750 | 1.093 | 0.776 | |
| r | 0.992 | 0.870 | 0.996 | 0.852 | |
| SE | 4.734 | 1.251 | 3.348 | 1.400 |

Conclusions
References
- Ikizler, A. A.; Ikizler, A.; Yıldırım, N. Antitumor activities of some 4,5-dihydro-1H-1,2,4-triazol-5-ones. Monatsh. Chem. 1991, 122, 557–33. [Google Scholar] [CrossRef]
- Ikizler, A. A.; Yüksek, H. Reaction of 4-amino-4,5-dihydro-1H-1,2,4-triazol-5-ones with 2,5-dimethoxytetrahydrofuran. Collect. Czech. Chem. Commun. 1994, 59, 731–735. [Google Scholar] [CrossRef]
- Bahçeci, Ş.; Yüksek, H.; Ocak, Z.; Azaklı, A.; Alkan, M.; Ozdemir, M. Synthesis and potentiometric titrations of some new 4-(benzylideneamino)-4,5-dihydro-1H-1,2,4-triazol-5-one derivatives in non-aqueous media. Collect. Czech. Chem. Commun. 2002, 67, 1215–1222. [Google Scholar] [CrossRef]
- Bahçeci, Ş.; Yüksek, H.; Ocak, Z.; Köksal, C.; Ozdemir, M. Synthesis and non-aqueous medium titrations of some new 4,5-dihydro-1H-1,2,4-triazol-5-one derivatives. Acta Chim. Slov. 2002, 49, 783–794. [Google Scholar]
- Yüksek, H.; Ocak, Z.; Özdemir, M.; Ocak, M.; Bekar, M.; Aksoy, M. A study on novel 4-heteroarylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-ones. Indian J. Heterocycl. Ch. 2003, 13, 49–52. [Google Scholar]
- Yüksek, H.; Bahçeci, Ş.; Ocak, Z.; Alkan, M.; Ermiş, B.; Mutlu, T.; Ocak, M.; Özdemir, M. Synthesis of some 4,5-dihydro-1H-1,2,4-triazol-5-ones. Indian J. Heterocycl. Ch. 2004, 13, 369–372. [Google Scholar]
- Ikizler, A. A.; Uçar, F.; Yüksek, H.; Aytin, A.; Yasa, I.; Gezer, T. Synthesis and antifungal activity of some new arylidenamino compounds. Acta Pol. Pharm.-Drug Res. 1997, 54, 135–140. [Google Scholar]
- Yüksek, H.; Demibaş, A.; Ikizler, A.; Johansson, C. B.; Çelik, C.; Ikizler, A. A. Synthesis and antibacterial activities of some 4,5-dihydro-1H-1,2,4-triazol-5-ones. Arzneim.-Forsch./Drug Res. 1997, 47, 405–409. [Google Scholar]
- Burzozowski, Z. Synthesis and anti-HIV activity of some new 2-mercapto-N-(1,2,4-triazol-3-yl)benzenesulfonamide derivatives containing the 1,2,4-triazole moiety fused with variety of heteroaromatic rings. Acta Pol. Pharm.-Drug Res. 1998, 55, 473–480. [Google Scholar]
- Bhat, A. R.; Bhat, G. V.; Shenoy, G. G. Synthesis and in-vitro antimicrobial activity of new 1,2,4-triazoles. J. Pharm. Pharmacol. 2001, 53, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, A. A.; Ikizler, A.; Yüksek, H.; Serdar, M. Antitumor activities of some 4,5-dihydro-1H-1,2,4-triazol-5-ones. Model. Measur. Control, Ser. C. 1998, 1, 25–33. [Google Scholar]
- Ikizler, A. A.; Demibaş, A.; Johansson, C. B.; Çelik, C.; Serdar, M.; Yüksek, H. Synthesis and biological activities of some 4,5-dihydro-1H-1,2,4-triazol-5-one derivatives. Acta Pol. Pharm.-Drug Res. 1998, 55, 117–123. [Google Scholar]
- Schleyer, P.V.R.; Allinger, N.L.; Clark, T.; Gasteiger, J.; Kolmann, P.A.; Schaefer, H.F.; Schreiner, P.R. (Eds.) The encyclopedia of computational chemistry; John Wiley & Sons: Chichester, 1998.
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. I A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Barfiled, M.; Fagerness, P. J. Density Functional Theory/GIAO Studies of the 13C, 15N, and 1H NMR chemical shifts in aminopyrimidines and aminobenzenes: relationships to electron densities and amine group orientations. J. Am. Chem. Soc. 1977, 119, 8699–8711. [Google Scholar] [CrossRef]
- Manaj, J. M.; Maciewska, D.; Waver, I. 1H, 13C and 15N NMR and GIAO CPHF calculations on two quinoacridinium salts. Magn. Reson. Chem. 2000, 38, 482–485. [Google Scholar] [CrossRef]
- Ikizler, A. A.; Un, R. Reactions of ester ethoxycarbonylhydrazones with some amine type compounds. Chim. Acta Turc. 1979, 7, 269–290, [Chem. Abstr. 1991, 94, 15645d]. [Google Scholar]
- Frisch, M. J.; et al. Gaussian 03, Revision C. 02; Gaussian, Inc.: Pittsburgh PA, 2003. [Google Scholar]
- Hameka, H. F. On the magnetic shielding in the hydrogen molecule. Mol. Phys. 1958, 1, 203–215. [Google Scholar]
- Wolinski, K.; Hilton, K. J. F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem.Soc. 1990, 12, 8251–8260. [Google Scholar]
- Womacott, T. M.; Womacott, R. J. Introductory Statistics, 5th edn; Wiley: New York, 1990. [Google Scholar]
- Poirer, R.; Kari, R.; Csizmadia, I.G. Handbook of Gaussian Basis Sets: A compendium for Ab Initio Molecular Orbital Calculations; Elsevier Science Publishing: New York, 1985. [Google Scholar]
- Forsyth, D A.; Sebag, A. B. Computed 13C-NMR Chemical Shifts via Empirically Scaled GIAO Shieldings and Molecular Mechanics Geometries. Conformation and Configuration from 13C Shifts. J. Am. Chem. Soc. 1997, 119, 9483–9494. [Google Scholar]
- Ikizler, A. A.; Ikizler, A.; Şentürk, H. B.; Serdar, M. The pKa values of some 1,2,4-triazole and 1,2,4-triazolin-5-one derivatives in nonaqueous media. Doğa-Tr. Kimya D. 1988, 12, 57–66, [Chem. Abstr. 1988, 109, 238277q]. [Google Scholar]
- Yüksek, H.; Alkan, M.; Ocak, Z.; Bahçeci, Ş.; Ocak, M.; Özdemir, M. Synthesis and acidic properties of some new potential biologically active 4-acylamino-4,5-dihydro-1H-1,2,4-triazol-5-one derivatives. Indian J. Chem. 2004, 43B, 1527–1531. [Google Scholar]
- Yüksek, H.; Ocak, Z.; Alkan, M.; Bahçeci, Ş.; Özdemir, M. Synthesis and determination of pKa values of some new 3,4-disubstituted-4,5-dihydro-1H-1,2,4-triazol-5-one derivatives in non-aqueous solvents. Molecules 2004, 9, 232–240. [Google Scholar]
- Yüksek, H.; Bahçeci, Ş.; Ocak, Z.; Özdemir, M.; Ocak, M.; Ermiş, B.; Mutlu, T. Synthesis and determination of acid dissociation constants of some new 4,5-dihydro-1H-1,2,4-triazol-5-one derivatives. Asian J. Chem. 2005, 17, 195–201. [Google Scholar]
- Ikizler, A. A.; Yüksek, H. A study on 4,5-dihydro-1H-1,2,4-triazol-5-ones. Rev. Roum. Chim. 1996, 41, 585–590. [Google Scholar]
- Ikizler, A. A.; Yüksek, H. Acylation of 4-amino-4,5-dihydro-1H-1,2,4-triazol-5-ones. Org. Prep. Proced. Int. 1993, 25, 99–105. [Google Scholar] [CrossRef]
- Ikizler, A. A.; Ikizler, A.; Yüksek, H. 1H NMR spectra of some 4,5-dihydro-1,2,4-triazol-5-ones. Magn. Reson. Chem. 1993, 31, 1088–1094. [Google Scholar] [CrossRef]
© 2005 by MDPI (http://www.mdpi.org).
Share and Cite
Yüksek, H.; Cakmak, I.; Sadi, S.; Alkan, M.; Baykara, H. Synthesis and GIAO NMR Calculations for Some Novel 4-Heteroarylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives: Comparison of Theoretical and Experimental 1Hand 13C- Chemical Shifts. Int. J. Mol. Sci. 2005, 6, 219-229. https://doi.org/10.3390/i6060219
Yüksek H, Cakmak I, Sadi S, Alkan M, Baykara H. Synthesis and GIAO NMR Calculations for Some Novel 4-Heteroarylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives: Comparison of Theoretical and Experimental 1Hand 13C- Chemical Shifts. International Journal of Molecular Sciences. 2005; 6(6):219-229. https://doi.org/10.3390/i6060219
Chicago/Turabian StyleYüksek, Haydar, Ismail Cakmak, Sibel Sadi, Muzaffer Alkan, and Haci Baykara. 2005. "Synthesis and GIAO NMR Calculations for Some Novel 4-Heteroarylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives: Comparison of Theoretical and Experimental 1Hand 13C- Chemical Shifts" International Journal of Molecular Sciences 6, no. 6: 219-229. https://doi.org/10.3390/i6060219