A Comparative X-ray Diffraction Study and Ab Initio Calculation on RU60358, a New Pyrethroid

Abstract
:1. Introduction

2. Results and discussion
2.1. Description of the crystal structure



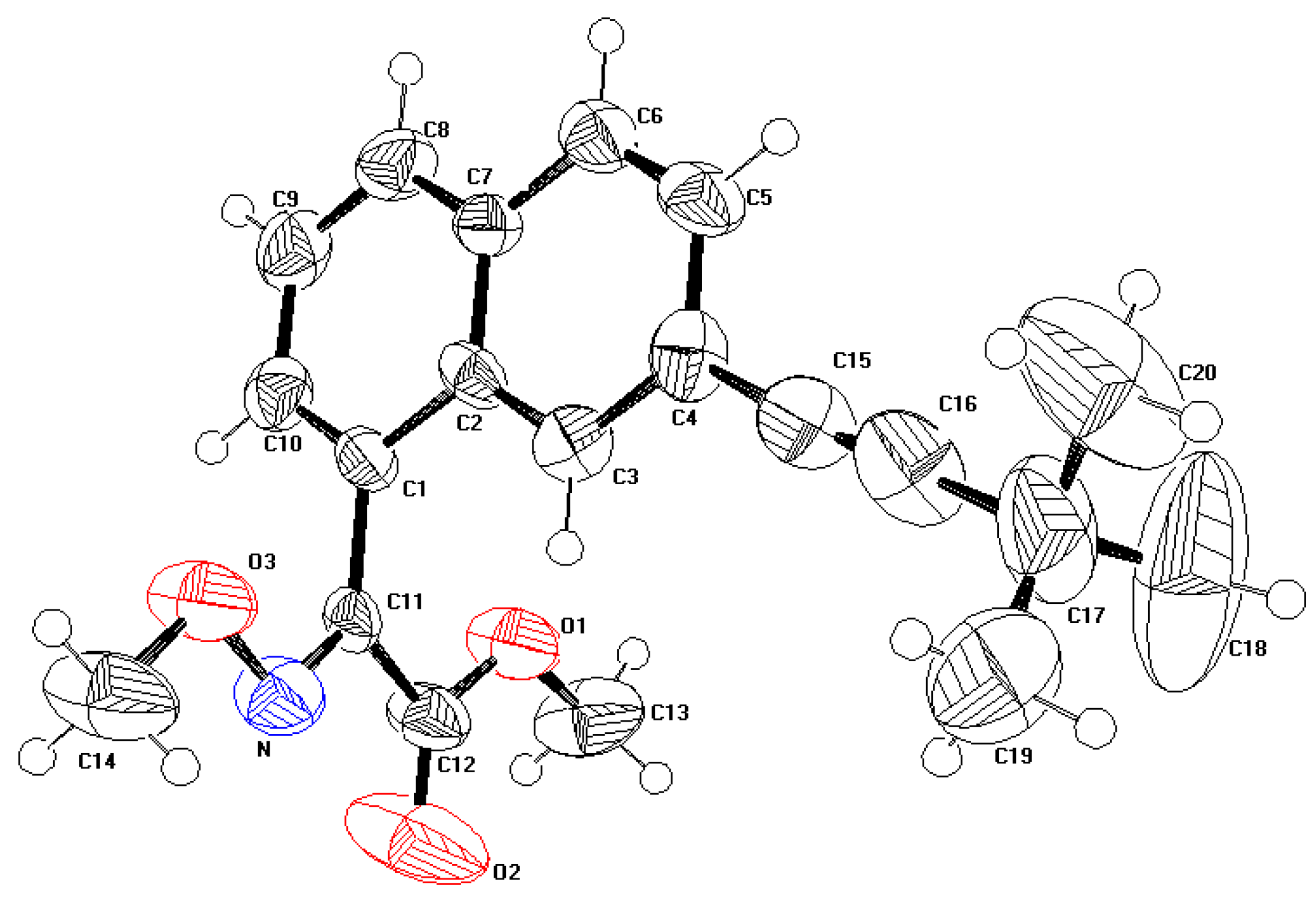{kind=link}
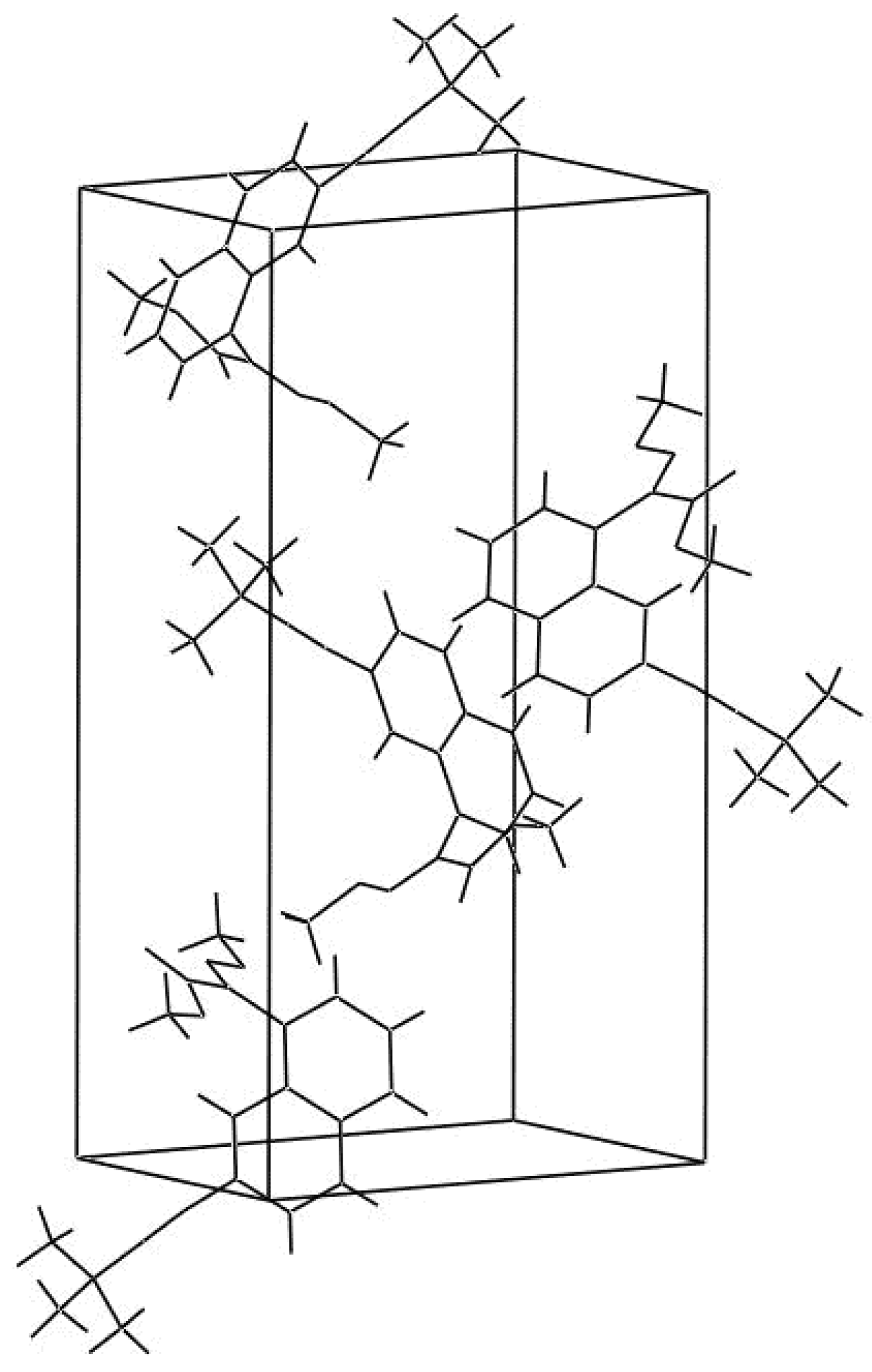{kind=link}
| Atom 1 | Atom 2 | Distance (Å) | |
|---|---|---|---|
| X-ray | B3LYP/6-31G** | ||
| C1 | C2 | 1.395 (10) | 1.433 |
| C1 | C10 | 1.395 (11) | 1.381 |
| C1 | C11 | 1.474 (12) | 1.472 |
| C2 | C3 | 1.410 (09) | 1.416 |
| C2 | C7 | 1.395 (11) | 1.433 |
| C3 | C4 | 1.395 (13) | 1.388 |
| C4 | C5 | 1.395 (10) | 1.428 |
| C4 | C15 | 1.429 (10) | 1.428 |
| C5 | C6 | 1.395 (10) | 1.371 |
| C6 | C7 | 1.395 (09) | 1.422 |
| C7 | C8 | 1.395 (11) | 1.418 |
| C8 | C9 | 1.395 (11) | 1.375 |
| C9 | C10 | 1.395 (09) | 1.412 |
| C11 | N | 1.278 (13) | 1.290 |
| C11 | C12 | 1.514 (15) | 1.497 |
| C12 | O1 | 1.292 (10) | 1.351 |
| C12 | O2 | 1.152 (10) | 1.210 |
| C13 | O1 | 1.439 (12) | 1.436 |
| N | O3 | 1.376 (09) | 1.374 |
| C14 | O3 | 1.427 (11) | 1.467 |
| C15 | C16 | 1.163 (13) | 1.213 |
| C16 | C17 | 1.467 (12) | 1.459 |
| C17 | C18 | 1.439 (12) | 1.548 |
| C17 | C19 | 1.469 (11) | 1.541 |
| C17 | C20 | 1.531 (13) | 1.549 |
| Atom 1 | Atom 2 | Atom 3 | Angle (°) | |
|---|---|---|---|---|
| X-ray | B3LYP/6-31G** | |||
| C2 | C1 | C10 | 120.0 (8) | 119.8 |
| C2 | C1 | C11 | 123.5 (5) | 121.1 |
| C10 | C1 | C11 | 116.2 (8) | 119.1 |
| C1 | C2 | C3 | 120.2 (6) | 122.6 |
| C1 | C2 | C7 | 120.0 (7) | 118.5 |
| C3 | C2 | C7 | 119.8 (7) | 118.7 |
| C2 | C3 | C4 | 120.0 (5) | 121.5 |
| C3 | C4 | C5 | 120.0 (5) | 119.2 |
| C3 | C4 | C15 | 122.1 (6) | 120.9 |
| C5 | C4 | C15 | 117.9 (8) | 119.9 |
| C4 | C5 | C6 | 120.0 (8) | 120.4 |
| C5 | C6 | C7 | 120.4 (7) | 121.2 |
| C2 | C7 | C6 | 119.8 (9) | 118.8 |
| C2 | C7 | C8 | 120.0 (5) | 119.6 |
| C6 | C7 | C8 | 120.2 (6) | 121.6 |
| C7 | C8 | C9 | 120.0 (5) | 120.6 |
| C8 | C9 | C10 | 120.0 (7) | 120.1 |
| C16 | C17 | C19 | 111.5 (9) | 109.4 |
| C16 | C17 | C20 | 107.7 (9) | 109.3 |
| C18 | C17 | C19 | 110.5 (8) | 109.7 |
| C18 | C17 | C20 | 107.3 (5) | 109.6 |
| C19 | C17 | C20 | 107.2 (4) | 109.5 |
| C1 | C10 | C9 | 120.0 (6) | 121.2 |
| C1 | C11 | N | 126.9 (8) | 125.7 |
| C1 | C11 | C12 | 121.6 (5) | 121.4 |
| N | C11 | C12 | 111.5 (4) | 112.9 |
| C12 | O1 | C13 | 117.0 (9) | 115.0 |
| N | O3 | C14 | 108.5 (5) | 109.2 |
| C11 | N | O3 | 110.8 (7) | 112.6 |
| C11 | C12 | O1 | 110.6 (7) | 110.5 |
| C11 | C12 | O2 | 126.0 (9) | 125.7 |
| O1 | C12 | O2 | 123.4 (5) | 123.8 |
| C4 | C15 | C16 | 174.8 (9) | 180.5 |
| C15 | C16 | C17 | 179.3 (6) | 180.1 |
| C16 | C17 | C18 | 112.6 (7) | 109.6 |
2.2. Optimized geometry
2.3. Vibrational wavenumbers
| ν (cm-1)* | Assignments | ν (cm-1)* | Assignments |
|---|---|---|---|
| 10.6 | τC17CT, τC1C11, δC4,C15,C16 | 702.1 | νCACA, νC1C11, δCACACA, δCT,O,N |
| 13.8 | τC1C11 | 723.4 | νsCT(CH3)3 (tBu) |
| 24.7 | τC11C12 | 738.1 | γCAH, γCACA, τCACA |
| 30.9 | δCA,C4,C15 | 749.6 | γC12=O2, γC11N, δC12=O2 |
| 35.4 | τC4C15, τC1C2, δC15,C16,C17 | 752.2 | γC12=O2, δC12=O2, δC12O1CT, νC12O1 |
| 61.9 | τC1C11, τC1CA, τC4C15 | 778.7 | γCAH, γC1C11, γCACA |
| 67.2 | τNO, γC11N | 819.0 | γCAH#, γCACA |
| 98.1 | τC14O, τNO | 825.3 | δCACACA(ring 1), γCAH##, νCACA |
| 102.4 | δC15,C16,C17, τC6C7 | 832.4 | δCACACA, νC16C17, νC17CT |
| 108.4 | δC12,C11,N, τO1C13, δC1,C11,C12 | 873.0 | γC3H, τ CACA |
| 112.8 | τO1C13 | 887.1 | γCAH(ring 1) |
| 129.4 | δC4,C15,C16, τO1C12 | 897.4 | νO3N, νO3CT, νO1C12, νO1CT,νC17CT |
| 158.9 | τC2C7, τC2C3, τC7C8 | 897.8 | νC17CT, ρ(tBu) |
| 163.6 | τO1C12, τO3C14, τON | 899.7 | νC17CT |
| 173.9 | τON, δC4,C15,C16, τO3C14 | 926.0 | νC17CT, νNO3, νC4CA, δCACACA |
| 185.2 | τC6C7, τC1C2, τC4C15 | 933.0 | γCAH( all rings) |
| 218.6 | νC4C15, νC16C17, δC3,C4,C5 | 933.8 | ρ(tBu) |
| 221.9 | τC17CH3 (tBu) | 942.9 | γCAH(all rings) |
| 246.0 | δC12,O1,C13, δO1,C12,C11, τC4C15 | 971.6 | νO3N, νCACA, δCACACA, δCAH, δC11,N,O3 |
| 269.5 | τC17CH3(tBu) | 996.9 | νCTO1, νO3CT |
| 276.5 | τC17CH3(tBu) | 1013.9 | ρ(tBu) |
| 279.4 | δC12,O1,C13, νC1C11, δC1,C11,C12 | 1014.1 | ρ(tBu) |
| 297.8 | τC11N, δC12,O1,C13, τO1C12 | 1045.1 | νCACA, νO3CT, νO3N |
| 314.3 | δC14,O,N, νC1C11, τC11N, δCACACA | 1060.2 | νO3CT, νCACA, νO3N |
| 332.4 | δCT,C17,CT(tBu) | 1095.2 | νC12O1, νCTO1, δC11,N,O3, νO3CT |
| 336.7 | δCT,C17,CT(tBu) | 1130.9 | ρC14H3 |
| 357.3 | δCT,C17,CT(tBu), δC1,C2,C3, δC6,C7,C8 | 1133.8 | ρC13H3 |
| 367.4 | νC11C12, δC11,N,O, δC12,C11,N | 1134.5 | δCAH, νCACA(ring 2) |
| 373.6 | δC11,C12,O2, δC12,O1,C13 | 1148.6 | δCAH, νCACA(ring 1) |
| 387.9 | τC4C15, τC4CA, γC6C7 | 1163.1 | ρC13H3 |
| 424.2 | δCT,C17,CT(tBu) | 1172.1 | ρC14H3 |
| 425.2 | τCACA, γCAH | 1181.6 | ρ(tBu) |
| 474.6 | δC4,C15,C16, δC16,C17,CT, γC-N | 1183.4 | ρ(tBu) |
| 501.5 | δC16,C17,CT, δCA,C4,C15, δCACACA | 1184.8 | νO1C12, ρC14H3, νC4C15, ρC13H3, ρ(tBu) |
| 512.7 | δC16,C17,CT, δC15,C16,C17, τCACA | 1196.4 | νCACA, δCAH |
| 528.1 | γCACA, τCACA, γC1C11, δC14,O3,N | 1215.3 | δCAH, νO1C12 |
| 545.3 | τC4C15, δC15,C16,C17, δC16,C17,CT,τCACA, γC4C15 | 1241.2 | δCAH(ring 2) |
| 550.4 | δC4,C15,C16, δC16,C17,CT | 1266.0 | νC11C12, νC1C11, νO1C12, δCAH(ring 1) |
| 583.3 | τC11N, νC16C17, δCA,C1,C11, γC11N, γC12=O2 | 1270.9 | νC16C17, νC4C15, δCAH(ring 1) |
| 590.2 | γC11N, τC11C12, γC12=O2, g C4C15 | 1303.8 | νCACA, δCAH |
| 616.1 | δCACACA, δCT,O,N, νC17CT | 1352.3 | νCACA |
| 652.7 | γC4C15, γC1C11, τCACA, γCAH | 1353.7 | dsCH3(tBu) |
| 1354.4 | dsCH3(tBu) | 2243.4 | νC15C16 |
| 1364.7 | νCACA, δCAH | 2925.2 | νsCH3 (tBu) |
| 1384.7 | δsCH3(tBu) | 2925.8 | νsCH3(tBu) |
| 1416.2 | δsC14H3 | 2931.6 | νsC14H3 |
| 1421.4 | δsC13H3 | 2931.8 | νsC14H3, nsCH3(tBu) |
| 1423.3 | δsC13H3 | 2947.1 | νsC13H3 |
| 1433.6 | δaCH3(tBu) | 2996.8 | νaCH3(tBu) |
| 1434.7 | δaC14H3 | 2997.4 | νaCH3(tBu) |
| 1436.2 | νCACA, δCAH | 3002.5 | νaCH3 (tBu) |
| 1437.8 | δaC13H3 | 3006.2 | νaC14H3 |
| 1443.9 | δaCH3 (tBu) | 3006.8 | νaCH3(tBu) |
| 1444.4 | δaCH3(tBu) | 3010.2 | νaCH3(tBu) |
| 1451.5 | δaC13H3 | 3010.9 | νaCH3(tBu) |
| 1456.3 | δaCH3(tBu) | 3020.7 | νaC13H3 |
| 1456.8 | δaCH3(tBu) | 3041.2 | νaC14H3 |
| 1457.4 | δaC14H3 | 3054.8 | νaC13H3 |
| 1474.6 | δaCH3(tBu) | 3056.3 | νCAH(all rings) |
| 1484.6 | νCACA, δCAH | 3059.4 | νCAH(all rings) |
| 1557.7 | νCACA, δCAH | 3068.8 | νCAH(ring 1) |
| 1582.1 | νC11N, νCACA | 3083.6 | νCAH(ring 1) |
| 1593.9 | νC11N, νCACA | 3087.9 | νCAH(ring 2) |
| 1609.1 | νCACA | 3096.9 | νCAH(ring 2) |
| 1749.0 | νC12=O2 |
3. Experimental Section
3.1 X-ray structure determination
3.2. Computational methods
| Crystal data | |
|---|---|
| Formula | C20H21NO3 |
| Molecular weight | 323.34 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell determination | Least-squares fit from 25 reflections (2° < q < 30°) |
| a (Å) | 7.7575 |
| b (Å) | 11.3182 |
| c (Å) | 21.3529 |
| V (Å3) | 1874.80 |
| Z | 4 |
| dcalc(g. cm-3) | 1.16 |
| mu (mm-1) | 0.077 |
| Crystal colour | Colourless |
| Crystal size | 0.32 ´ 0.27 ´ 0.10 ( mm) |
| Experimental data | |
| Technique | Four circles diffractometer, CAD4 Enraf Nonius kappa geometry |
| Graphite oriented monochromator : Moka | |
| λ = 0.71070 Å, ω/2q scan | |
| Scanning range for θ | 2.04 ¾ 29.96 |
| Number of reflections measured | 3082 |
| Number of reflections observed | 1583 (I ≥ 3σ(I)criterion) |
| Limiting indices | h 0 ® 10 |
| k 0 ® 15 | |
| l 0 ® 30 | |
| T (K) | 293 |
| Refinement data | |
| Refinement method | Full-matrix least-squares on F |
| Final R indices | R=0.068, wR=0.068 |
| S | 1.249 |
| H atoms | constrained refinement |
| Parameters | 224 |
| (Δ/σ)max | 0.380 |
| (Δρ)min | -0.271 eÅ-3 |
| (Δρ)max | 0.380 eÅ-3 |
| Extinction | No extinction correction applied |
References
- Baert, F.; Guelzim, A. X-ray Structure of the Pyrethroid Insecticide { 1R-[1 α(S*),2α] }-2-(2,2-Dichlorovinyl)-3,3-dimethylcyclopropanecarboxylic Acid Cyano(3-phenoxyphenyl)methyl Ester (RU 24501). Acta Cryst. C 1991, 47, 606–608. [Google Scholar] [CrossRef]
- Baert, F.; Guelzim, A.; Germain, G. Structure of Two Pyrethroid Insecticides: Acrynathryn (RU 38702) and a Derivative (RU 38181). Acta Cryst. C 1991, 47, 768–771. [Google Scholar] [CrossRef]
- Hamzaoui, F.; Lamiot, J.; Baert, F. X-ray Structure of a New Pyrethroid RU 52259. Acta Cryst. C 1993, 49, 818–820. [Google Scholar] [CrossRef]
- Hamzaoui, F.; Baert, F. A New Pyrethroid Insecticide RU41414. Acta Cryst. C 1996, 52, 689–690. [Google Scholar] [CrossRef]
- Tessier, J. Recent Advances in the Chemistry of Insect Control; Janes, N.F., Ed.; The Royal Society of Chemistry: London, 1985; pp. 26–52. [Google Scholar]
- Tessier, J.; Teche, A.; Demoute, J. P. Pesticide Chemistry: Human Welfare and the Environment; Miyamoto, J., Kearney, P.C., Eds.; Pergamon Press: Oxford, New York, 1983; Vol. 1, pp. 95–100. [Google Scholar]
- Tessier, J.; Teche, A.; Demoute, J. P. Proceedings of the 5th IUPAC International Congress of Pesticide Chemistry; Miyamoto, J., Kearney, P.C., Eds.; Pergamon Press: Oxford, New York, 1983; pp. 197–202.
- Babin, D.; Demassey, J.; Demoute, J. P.; Dutheil, P.; Terrie, I.; Tessier, J. A New Way toward Zα,β Unsaturated Esters: A Pyrethroid Application. J. Org. Chem. 1992, 57, 584–589. [Google Scholar] [CrossRef]
- Elliott, M. The Relationship between the Structure and the Activity of Pyrethroids. Bull. Wld Hlth Org. 1970, 44, 315–324. [Google Scholar]
- Elliott, M.; Farnham, A. W.; Janes, N. F.; Needham, P. H.; Pulman, D. A. Insecticidal Activity of the Pyrethrins and Related Compounds. Pestic. Sci. 1975, 6, 537–542. [Google Scholar] [CrossRef]
- Allouche, A.; Pourcin, J. Ab initio calculation of vibrational force fields: Determination of non-redundant symmetry coordinates by least-square component analysis. Spectrochim. Acta, Part A 1993, 49, 571–580. [Google Scholar] [CrossRef]
- Pulay, P. Possibilities and limitations of ab initio calculation of vibrational spectra. J. Mol. Struct. 1995, 347, 293–308. [Google Scholar] [CrossRef]
- Dollish, F. R.; Fateley, W. G.; Bentley, F. F. Characteristic Raman Frequencies of Organic Compounds; John Wiley& Sons: New York, 1974. [Google Scholar]
- Varsányi, G.; Szöke, S. Vibrational Spectra of Benzene Derivatives; Academic Press: New York, 1969. [Google Scholar]
- Wilson, E. B., Jr. The Normal Modes and Frequencies of Vibration of the Regular Plane Hexagon Model of the Benzene Molecule. Phys. Rev. 1934, 45, 706–714. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS97: Program for crystal structure determination; University of Göttingen: Germany, 1997. [Google Scholar]
- Sheldrick, G. M. SHELXL97: Program for crystal structure determination; University of Göttingen: Germany, 1997. [Google Scholar]
- Brown, P. J.; Fox, A. G.; Maslen, E. N.; O'Keefe, M. A.; Willis, B. T. M. International Tables for X-ray Crystallography; Wilson, A. J.C., Prince, E., Eds.; Kluwer Academic Publishers: Dordrecht, 1999; Vol. C, pp. 548–589. [Google Scholar]
- Stewart, R. F.; Davidson, E. R.; Simpson, W. T. Coherent X-Ray Scattering for the Hydrogen Atom in the Hydrogen Molecule. J. Chem. Phys. 1965, 42, 3175–3187. [Google Scholar] [CrossRef]
- Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Rauhut, G.; Pulay, P. Transferable scaling factors for density functional derived vibrational force fields. J. Phys. Chem. 1995, 99, 3093–3100. [Google Scholar] [CrossRef]
- Gómez Marigliano, A. C.; Varetti, E. L. Self-Association of Formamide in Carbon Tetrachloride Solutions: An Experimental and Quantum Chemistry Vibrational and Thermodynamic Study. J. Phys. Chem. A 2002, 106, 1100–1106. [Google Scholar] [CrossRef]
- Gómez Marigliano, A.C.; Varetti, E.L. Unpublished results.
- Sim, F.; St.-Amant, A.; Papai, I.; Salahub, D. R. Gaussian density functional calculations on hydrogen-bonded systems. J. Am. Chem. Soc. 1992, 114, 4391–4400. [Google Scholar] [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision A.1. Gaussian, Inc.: Pittsburgh PA, 2003. [Google Scholar]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Hamzaoui, F.; Chouaih, A.; Lagant, P.; Belarbi, O.; Vergoten, G. A Comparative X-ray Diffraction Study and Ab Initio Calculation on RU60358, a New Pyrethroid. Int. J. Mol. Sci. 2006, 7, 255-265. https://doi.org/10.3390/i7080255
Hamzaoui F, Chouaih A, Lagant P, Belarbi O, Vergoten G. A Comparative X-ray Diffraction Study and Ab Initio Calculation on RU60358, a New Pyrethroid. International Journal of Molecular Sciences. 2006; 7(8):255-265. https://doi.org/10.3390/i7080255
Chicago/Turabian StyleHamzaoui, Fodil, Abdelkader Chouaih, Philippe Lagant, Ouassila Belarbi, and Gérard Vergoten. 2006. "A Comparative X-ray Diffraction Study and Ab Initio Calculation on RU60358, a New Pyrethroid" International Journal of Molecular Sciences 7, no. 8: 255-265. https://doi.org/10.3390/i7080255