A Fragment-Based Scoring Function to Re-rank ATP Docking Results

1
M.M. Shemyakin & Yu.A. Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Ul. Miklukho-Maklaya, 16/10, 117997 GSP, Moscow V-437, Russia
2
Moscow Institute of Physics and Technology (State University), 141700, Institutskii per., 9, Dolgoprudny, Moscow region, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2007, 8(11), 1083-1094; https://doi.org/10.3390/i8111083
Submission received: 3 August 2007
/
Revised: 29 September 2007
/
Accepted: 22 October 2007
/
Published: 31 October 2007
(This article belongs to the Section Molecular Recognition)
Abstract
:ATP is involved in numerous biochemical reactions in living cells interacting with different proteins. Molecular docking simulations provide considerable insight into the problem of molecular recognition of this substrate. To improve the selection of correct ATP poses among those generated by docking algorithms we propose a post-docking reranking criterion. The method is based on detailed analysis of the intermolecular interactions in 50 high-resolution 3D-structures of ATP-protein complexes. A distinctive new feature of the proposed method is that the ligand molecule is divided into fragments that differ in their physical properties. The placement of each of them into the binding site is judged separately by different criteria, thus avoiding undesirable averaging of the scoring function terms by highlighting those relevant for particular fragments. The scoring performance of the new criteria was tested with the docking solutions for ATPprotein complexes and a significant improvement in the selection of correct docking poses was observed, as compared to the standard scoring function.
1. Introduction
ATP participates in a great variety of biological processes. It is involved in cell signaling by phosphorylating proteins and serves as a universal carrier of energy. Disfunction of numerous proteins – ATP-targets – can cause severe diseases and other abnormalities in humans [1–3]. Therefore, understanding the principles of ATP-protein recognition is necessary for investigation of enzymatic mechanisms. While the number of proteins with known 3D structure is rapidly increasing, structural characterization of protein-ligand complexes is still experimentally demanding. This raises the problem of efficient ways to determine the conformations of such complexes by in silico techniques. Molecular docking is the most common way to treat this problem.
A number of different docking softwares have been proposed to solve the docking problem, all of them providing more or less precise predictions of ligand binding to a target (see [4,5] for reviews). In general, the docking process consists of a search algorithm and a scoring function. While both these components are being continuously improved, they still have some important limitations. Particularly, inaccuracy of scoring functions hinders delineation of correct solutions among those yielded by the docking run. One way to overcome this problem is to use more efficient post-docking filters [6]. Usually, these are target-oriented scoring functions [7] that include restraints such as a requirement for a potential ligand to form hydrogen bond(s) with a certain residue. These restraints are based on the knowledge of the structure and distinctive features of the active site of the protein under study. However, the improvement of the scoring procedure can also be introduced from the opposite side – by developing ranking criteria designed for a particular class of ligand molecules, as was done for carbohydrates [8], peptides [9], ATP [10].
In this work we describe the further improvement of the ATP-oriented scoring function proposed earlier [10]. While the previous version was focused only on estimation of the position of an adenine ring in the active site, here we have extended it on the whole molecule. To design a ligand-oriented scoring function one needs to know what intermolecular contacts are typical for complexes of this molecule with proteins. In case of ATP a considerable amount of such data has been already collected in numerous studies, thus making possible construction of the ATP-oriented score. For example, it was found that recognition of the phosphate groups (often with coordinated magnesium ions) is often driven by well-known phosphate-binding sequence motifs, such as Walker A [11], Kinase-1 and Kinase-2 [12], and the P-loop [13]. Intermolecular interactions of ribose also have been recently investigated indicating that hydrogen bonds between its hydroxyl groups and protein are often mediated by water molecules in ATP-, ADP-, and FAD-protein complexes [14]. Protein complexes with ATP, ADP, CoA, NAD, and FAD were studied thoroughly to identify the intermolecular interactions that drive the adenine binding [15–19]. According to these investigations, the most relevant is the adenine-protein hydrophobic contact. Taking this into account, special attention was paid to quantitative characterization of spatial hydrophobic/hydrophilic properties of ATP and its protein environment. Meanwhile, π-π stacking and cation-π interactions between the adenine base and surrounding aromatic residues, along with hydrogen bonds of the adenine amino group, also play an important role.
While most scoring functions are represented by a sum of different interaction terms over the whole ligand molecule, in the present work we propose a different approach. The total score is composed of scores for different fragments of the ligand molecule. For ATP, for example, these are the adenine ring and the phosphate tail. The idea was inspired by the pharmacophore method (see [20, 21] for reviews), in which molecules are divided into several fragments based on their physicochemical properties. In our method the placement of each of them is judged separately by different criteria thus, avoiding undesirable averaging of the scoring function terms, while highlighting those relevant for each particular fragment. The scoring performance of the proposed criteria demonstrates a significant improvement in the selection of correct docking poses as compared to the standard scoring function.
2. Results and Discussion
2.1 ATP docking and scoring
ATP docking was performed for 50 ATP-protein complexes (see Methods) and the generated ligand positions were compared with the experimental X-ray structures. The correctness of a solution was judged based on a cut-off of root-mean-square deviation (RMSD) from the reference structure. Thus, for the adenine and ribose moieties the cut-off value was set to 2.0 Å, while for the phosphate tail – to 3.0 Å, and for the whole ATP molecule – to 3.5 Å, taking into account the greater flexibility of the two latter cases. Applying these criteria to the results of docking demonstrated that both correct and wrong orientations of adenine and the whole ATP could be found for all 50 complexes, while for both ribose and phosphate tail there were only 46 such complexes. The fact that correct ATP positions were found in each of 50 complexes should not be considered to be in conflict with, for example, the absence of correct phosphate tail orientations in four complexes. The explanation is that the RMSD criterion applied to the whole molecule is more tolerant and, additionally, RMSD was averaged over the whole structure.
The fact that analysis of docking results for all 50 complexes revealed that in most cases both correct and wrong solutions were obtained indicates high efficiency of the ligand conformational search. Now, when we have in our hands quite a large and representative set of different docking solutions for ATP one needs a proper scoring method to select the correct ones among the majority of misleading positions. Scoring by the standard function “goldscore” [24] ranked correct ATP positions on top only for 22 out of 50 complexes (Table 1). To some extent such a poor hit rate (< 50%) can be explained by unusual docking parameters. Namely, the “goldscore” was trained on protein-ligand complexes including also coordinated metal ions, while in our study these ions were compulsorily removed, thereby distorting the “goldscore” values. Nevertheless, it would still be desirable to develop a new and more efficient ranking criterion to rank the results of ATP docking.
2.2 Design of fragmental scores
Moreover, taking into account our previous advantages in developing individual scoring criterion for the adenine moiety, it would also be interesting to see whether it is possible to design a fragmental scoring function. While in case of ATP it is easy to divide the molecule into parts differing in physical properties guided by intuitive considerations, it would also be desirable to elaborate some rigorous criteria for such a procedure. We propose to divide a molecule into fragments, or groups of atoms, based on their distinct hydrophobic/hydrophilic properties. It is rather easy to do using the Molecular Hydrophobicity Potential (MHP) approach [22], when each atom has its own value of hydrophobicity (or hydrophilicity, if negative). After that atoms are clusterized by these values. In this study we applied a cut-off value of 0.2 MHP units to judge whether two atoms bound by a covalent bond belong to the same cluster. However this procedure is not perfect yet, and needs further substantial improvement. Thus, for the sake of better description of a molecule in case of ATP we had to compulsory fuse the hydrophilic amino group with the hydrophobic adenine cluster, which would otherwise remain as independent fragment.
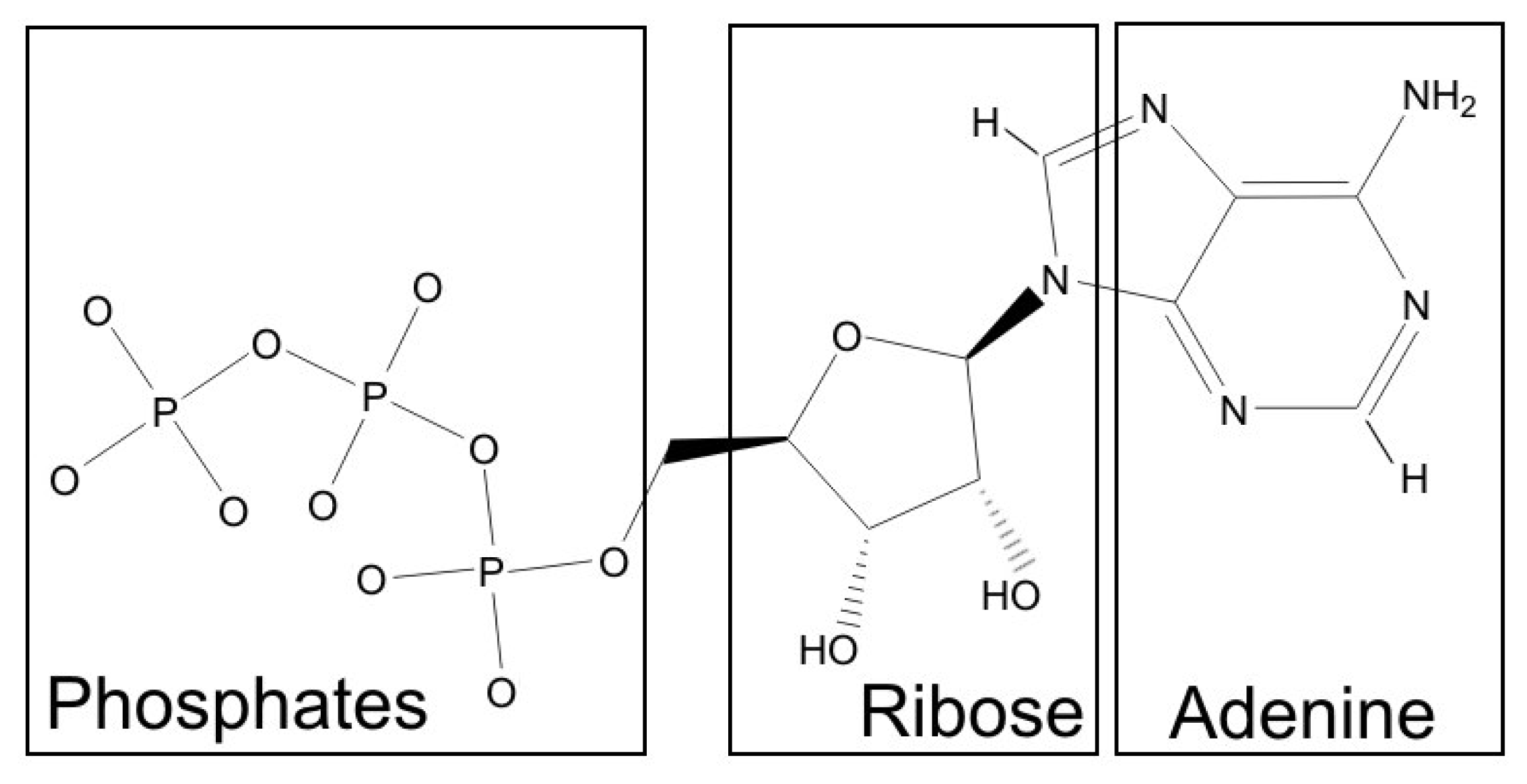
According to the procedure of dissecting a molecule into fragments, three fragments were identified in ATP. These corresponded well to the intuitively recognized parts with different physicochemical properties: the hydrophobic adenine ring, a slightly hydrophilic ribose moiety and a distinctly hydrophilic phosphate tail (Figure 1). It should be noted, however, that the “ribose fragment” includes also a part of the adenine ring, and the hydrophobic CH2 group separating ribose from phosphates was not included into any fragment. For each of these fragments, an individual scoring function was then constructed. With this aim, according to our previous results, we decided to seek for the new criteria combining hydrophobic/hydrophilic complementarity, hydrogen bonds, and aromatic interactions.
New scoring functions were designed as linear combinations of the corresponding interaction terms. The weighting coefficients of the terms were adjusted by the least-squares fitting procedure based on docking results. In the regression procedure, each scoring criterion was fitted to a switch function so as to yield the value of 1 in case of correct prediction and the value of 0 otherwise. For that purpose all fragment poses were divided into three groups corresponding to different values of the switch function. Correct structures, characterized by RMSD from the reference within 2.0 Å for adenine and ribose and 3.0 Å for the phosphate tail, were assigned the value of 1. Since it was hard to judge whether the poses with RMSD greater than the correctness threshold but less then 5.0 Å are correct or not, they were assigned an intermediate value of 0.5. Finally, all positions with the greater RMSD were assumed incorrect and therefore corresponding to the value of 0.
It is also important to note that the final formulas of the fragmental scores comprise different interaction terms and not all terms were included into each equation. This needs a more detailed explanation. Starting with the term that had the best hit rate alone, other terms were added subsequently until the increase of correlation between the score and the switch fitness function was greater than 0.01. According to the data presented in Table 1, hydrophobic complementarity was chosen as the starting point for adenine and ribose, and for phosphates – different acceptor hydrogen bonds and hydrophilic complementarity (the last being the most successful run). Such approach left only those terms that are really relevant for discriminating the correct solutions. This is necessary because otherwise including minor terms into the equation could lead to an overfitting effect, i.e. the performance of the proposed score will increase for the training set in exchange for decreasing in other cases. The resulting criterion for adenine includes the terms of hydrophobic complementarity, hydrogen bonds with the amino group and stacking and pi-cation interactions with Arg. This agrees well with our previous results [10]. The equation describing this criterion is (definitions of the terms are given in Methods):
The corresponding equations for the ribose and phosphate fragments are:
In all these equations the names of interaction terms correspond exactly to the terms in Table 1. Surprisingly, the scores for ribose and phosphates do not include any hydrogen bond term. This may seem confusing at first glance, but to all appearance in case of phosphates these bonds are implicitly taken into account by the hydrophilic interactions terms.
The performance of the proposed scores is represented in Table 1. Since the values of “goldscore” are related not to parts but to the whole ATP molecule, it is not quite correct to compare them with the fragmental scores, however it still can serve as a plausible reference point. It is clearly seen that the ADE_score and PHOS_score perform better than the “goldscore” while the RIB_score demonstrates very poor results.
Still, additional testing is required to make sure that overfitting was avoided. A common way to do that is to divide all the cases into two sets: the training and the test ones. However, still there is no generally accepted approach to perform such a division. So, in this study we propose to do that in the following way. Since overfitting is due to minor peculiarities of the training set, both sets should differ in minor features. At the same time, they should be similar in general properties like the strength of hydrophobic and/or hydrophilic interactions. To achieve this, all three sets were divided into two groups of an equal size by ranking them with the value of typical interactions observed in the X-ray structures (MHPphob-phob for adenine and MHPphil-phil for both ribose and phosphate tail). After that they were counted off by twos to create two sets. This procedure yielded similar distribution of hydrophobic/hydrophilic properties in both sets while being composed of different proteins they would likely differ in minor details such as the number of hydrogen bonds, etc.
These sets were then used in a cross test, where the weighting coefficients were trained with one set and then tested with another, and vice versa. The results are summarized in Table 2. It is clearly seen that the relative errors of the weighting coefficients of the ADE score are acceptable, while those of the PHOS_score are a bit worse. Nevertheless, this is quite a good result, since exclusion of one half of the training cases is rather a rigorous test. Furthermore, both these scores demonstrate almost the same hit rate, which is definitely higher than that of the “goldscore”. These results indicate that the values of the weighting coefficients in the both equations lie in admissible ranges, and the values obtained with the full sets will be further used (equations 1 and 3). The results for the RIB_score are completely unacceptable with very high relative error (> 0.70) of some coefficients. The last result is not surprising considering its relatively low hit rate (Table 1).
In summary, efficient scoring criteria were designed to select correct poses of the adenine ring and the phosphate tail. Unlike that, our approach did not yield any improvement in ranking for the ribose moiety. The reason for that is not quite clear and would require additional investigation. However, one may speculate that ribose often does not play significant role in ATP recognition, but serves as a linker between adenine and phosphates. This effect has been identified in some complexes of ATP and its analogues with proteins [3].
2.3 Performance of fragmental scores
Considering the quite encouraging improvement of ranking of ATP fragments it is interesting now to determine whether the same approach can be applied to the whole molecule. The same procedure was used in construction of a scoring criterion for the whole ligand molecule (results not shown). However, it exhibited only modest improvement as compared to “goldscore” increasing hit rate from 22 to 24, i.e. just by 4%.
Realizing that this methodology cannot yield sufficient improvement in scoring we decided to use another approach to construct a ranking criterion. Since the fragmental scores efficiently select correctly placed parts of ATP, the most obvious way was to combine them into an integral ranking function ATP_score. The best results were obtained for a mere sum of scores for adenine and phosphate – ADE_score and PHOS_score (see equation 4) – increasing the hit rate for ATP up to 35 out of 50, which is 26% better than the “goldscore”. Interestingly, addition of the RIB_score greatly spoils the ranking.
The presented results allow a conclusion that a scoring function composed of individual scores of molecular fragments performs considerably better than the one based on intermolecular interactions of the whole molecule. Thus, the fragmental scores highlight those interactions that really determine their recognition by the receptor. At the same time, other interactions that could distort the resulting picture, are attenuated or excluded at all. For example, comparison of the ADE_score and PHOS_score demonstrates that MHPphil-phil term is important for recognition of phosphates. Meanwhile, in case the ATP molecule is not dissected into fragments, the value of MHPphil-phil is calculated over the whole ligand including the ribose ring and adenine amino group. In the last case it becomes unclear whether this term remains useful for scoring any longer.
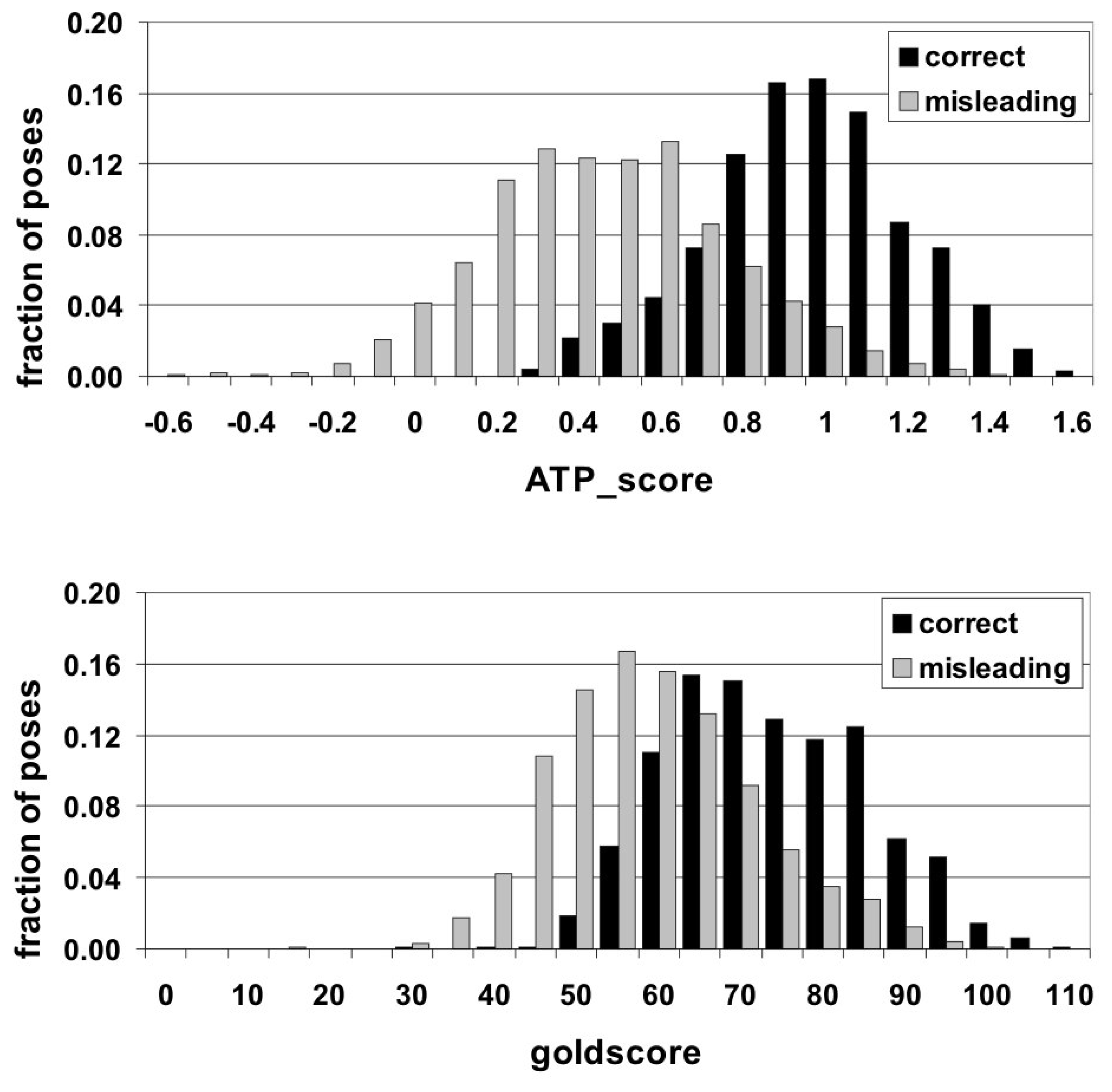
To summarize, it would be useful to present the results on the overall performance of the ATP_score. Besides the improved hit-rate, it provides better separation between correct and misleading poses. To illustrate this, two types of ATP poses of all 50 complexes were considered - correct (RMSD < 3.5 Å, 692 cases) and definitely incorrect (RMSD > 5.0 Å, 1851 cases) solutions. Other solutions (between these RMSD cut-offs) lie in the twilight zone where it is hard to understand whether such RMSD is caused by slight displacement of the whole molecule or by completely incorrect orientation of its part.
For all docking solutions the “goldscore” values fell into the range between 0 and 110 (in goldscore units). Theoretically, the ATP_score values should cover the interval between 0 and 2 (since in an ideal perfect case both ADE_score and PHOS_score are equal to 1). In practice, due to non-ideal interactions and inaccuracies of the fitting procedure they fell between −0.6 and 1.6. Comparing the distributions of both correct and misleading solutions over the values of the scoring functions (Figure 2), it is clearly seen that they are quite similar for “goldscore”, whereas ATP_score efficiently separates them. Numerically, this conclusion can be supported by the χ2 test yielding the values of 0.544 and 1.031 for “goldscore” and ATP_score, respectively. These results indicate that there can be proposed an ATP_score threshold for delimiting correct and misleading poses. The value ATP_score = 0.7 can be chosen as such a threshold since 90% of correct solutions have higher scores and 75% of misleading ones reveal lower values.
Similar distributions of correct and misleading positions were obtained for fragmental scoring functions for adenine and phosphates. It was found that the optimal ADE_score and PHOS_score cutoff values that separate correct and wrong solutions, are similar for both criteria and equal to 0.4 score units. The presented new ATP-oriented scoring function can be used to improve the results of docking ATP to different protein targets. It efficiently identifies those docking poses that are correctly oriented in the binding site. Further investigation will be aimed at improvement of the proposed score, e.g. considering interactions with metal ions, and development of similar approaches for a broader class of ligands.
3. Conclusions
A new improved scoring function is proposed for efficient selection of docked ATP poses. The new criterion is based on a fragmental approach, i.e. the total value of the score is computed as a combination of individual scores for different parts of the ligand. The advantage of such method as compared to the whole molecule approach was clearly demonstrated by ranking the results of ATP docking to 50 different protein targets.
Furthermore, one more advantage of the fragmental score is that it evaluates correctness of placement of individual parts of a ligand molecule. This can be useful taking into account considerable flexibility of some ligands and therefore their possibility to be docked “partly” correctly. In such case the fragmental scores help identify which exactly parts of ligand are in most favorable environment and which are not. While it remains to be seen, we hope that such information could also be used in rational design of ligands.
In the present work we also describe a methodology of construction of a fragmental scoring function. In future, it could be applied for development of fragment-based scoring functions for other ligands or classes of ligands. However, as seen from the example of the ribose moiety of ATP, there still remains a problem of identification of those fragments that drive the ligand recognition by receptor and therefore should be included into the integral scoring function and those that should be omitted.
4. Methods
4.1 ATP docking
ATP-protein complexes used in the present study were taken from the Protein Data Bank (PDB) [23] and are the same as in our previous work [10]. All of them belong to different protein classes and their atomic coordinates were defined by X-ray diffraction with resolution 3.0 Å or better. Docking simulations were performed with the GOLD 2.0 software [24] with standard parameters. The radius of the docking sphere was 30.0 Å, the origin being placed in the vicinity of the binding site. Prior to docking all water molecules were removed. Metal ions were also excluded to make the new scoring functions applicable to those cases where the correct position of a metal ion is not known (e.g. protein structures modeled by homology or extracted from molecular dynamics simulations). The X-ray structures were taken as they are without minimization. Since removal of positively charged metal ions could cause artifacts in electrostatic interactions we did not consider them in the present study.
4.2 Hydrophobic interactions
To estimate molecular hydrophobic/hydrophilic properties we used the Molecular Hydrophobicity Potential (MHP) formalism [22]. The atomic hydrophobicity constants were taken from the work by Ghose et al. [25]. In the MHP method these constants are assigned to atoms based on the topology of a molecule. Positive constants correspond to hydrophobic properties and negative – to hydrophilic ones.
MHP was used to evaluate the hydrophobic/hydrophilic complementarity of a ligand and its environment in the complex. The complementarity was divided into four types which were calculated separately – contacts of hydrophobic parts of ligand with hydrophobic (MHPphob-phob) and hydrophilic (MHPphob-phil) environment, and contacts of hydrophilic parts of ligand with hydrophobic (MHPphil-phob) and hydrophilic (MHPphil-phil) environment. All these values were calculated as the sums of the number of atom-atom contacts within 6 Å over all atoms. The contacts that fell between 5 and 6 Å were scaled linearly from 1 to 0.
To avoid artifacts that could arise from the absence of explicit water molecules in docking, a ligand molecule was surrounded by a rectangular grid, which imitated the hydrophilic properties of water. In calculation of MHP complementarity, the nodes of the grid were treated as atoms. The following parameters of the grid were used: spacing in all directions 2.0 Å, MHP constant of a node equal to – 0.38 MHP units.
Similarly, the embedding of an atom was calculated as the ratio of the sum of atom-atom contacts with the receptor to the sum of all atom-atom and atom-grid contacts.
4.3 Hydrogen bonds and stacking
Hydrogen bonds were identified by geometric criteria. All hydrogen bonds were treated equally and the corresponding interaction terms were calculated as simple counts of such bonds. Thus, a fullstrength hydrogen bond was assumed to be formed if the distance between the donor (D) and the acceptor (A) heavy atoms (rAD) was less than 3.4 Å and the angle ∠ADH (“H” denotes the hydrogen atom) is less than 50º. The hydrogen bond weight was linearly scaled to zero with the increase of the rAD up to 4.4 Å, or increase of the angle ∠ADH up to 65°.
Furthermore, the docking program GOLD [24] may introduce ambiguities in the positioning of hydrogen atoms of hydroxyl- and amino-groups of the receptor by adjusting them to form optimal hydrogen bonds. In this case to identify hydrogen bonds based on the initial protein structure we used another angle ∠CDA as a criterion. Here the symbol “C” denotes a heavy atom next to the atom D (in proteins this would usually be a carbon atom). The angle ∠CDA was assumed to be optimal in the range 105° ± 50°, scaling linearly to zero when reaching the values of 105° ± 65°. These values form a cone of possible positions of a hydrogen atom.
To identify which particular types of hydrogen bonds are important for ATP recognition by proteins, all such bonds were divided into six classes: charged-charged, charged-neutral, and neutralneutral formed by ligand donor (HBdcc, HBdcn, HBdnn) or acceptor (HBacc, HBacn, HBann). Such differentiation of hydrogen bonds was prompted by the analysis of other, publicly available scoring functions, in particular the Glide score XP [26] and by general considerations on the nature of hydrogen bond interactions.
Stacking interactions between the flat π conjugated rings of the adenine and those of Phe, Tyr, Trp and His, as well as the guanidine group of Arg (denoted in text as Pi-cation) were also identified by geometric criteria. For that purpose we introduced three weighting parameters, describing the angle α between the planes of these moieties, and the distance between their centers along the normal to the adenine plane (dnormal) and the relative displacement (dparallel – the distance between their centers along the adenine plane). The strength of the stacking interaction was then computed by considering these three geometrical descriptors:
The optimal value of 1 for S2(dnormal) was assigned when dnormal < 4.5 Å, scaled to zero up to dnormal = 5.5 Å. Similarly, the value of 1 for S3(dparallel) was assigned when dparallel <1.25 Å, scaled to zero up to dparallel = 2.50 Å. The angle function S1(α) is optimal when α = 0° and has the functional form of:
Figure 1.
Dissection of the ATP molecule into fragments based on atomic hydrophobic/hydrophilic properties. The fragments identified by an automated procedure correspond well to the intuitively distinguished parts: a hydrophobic adenine ring with the attached amino group, a slightly hydrophilic ribose moiety and a distinctly hydrophilic phosphate tail.
Figure 1.
Dissection of the ATP molecule into fragments based on atomic hydrophobic/hydrophilic properties. The fragments identified by an automated procedure correspond well to the intuitively distinguished parts: a hydrophobic adenine ring with the attached amino group, a slightly hydrophilic ribose moiety and a distinctly hydrophilic phosphate tail.

Figure 2.
Distribution of correct and misleading ATP docking solutions over the values of ATP_score (top) and “goldscore” (bottom).
Figure 2.
Distribution of correct and misleading ATP docking solutions over the values of ATP_score (top) and “goldscore” (bottom).


{kind=link}
{kind=link}
| Interaction term* | Adenine | Ribose | Phosphates | ATP |
|---|---|---|---|---|
| Total cases | 50 | 46 | 46 | 50 |
| “goldscore”** | 21 | 20 | 18 | 22 |
| NEW SCORE | 31 | 12 | 23 | 35 (24)*** |
| MHPphob-phob | 21 | 7 | 0 | 19 |
| MHPphob-phil | 1 | 1 | 0 | 7 |
| MHPphil-phob | 0 | 0 | 6 | 5 |
| MHPphil-phil | 10 | 6 | 15 | 10 |
| HBdcc | 0 | 0 | 0 | 0 |
| HBdcn | 1 | 6 | 0 | 4 |
| HBdnn | 11 | 3 | 0 | 10 |
| HBacc | 0 | 0 | 14 | 12 |
| HBacn | 1 | 1 | 16 | 13 |
| HBann | 2 | 5 | 15 | 8 |
| Embedding | 12 | 1 | 12 | 13 |
| Stacking | 13 | 0 | 0 | 11 |
| Pi_cation | 5 | 0 | 0 | 7 |
*the following abbreviations were introduced to denote interaction terms (see Methods for more detail): MHPphob-phob and MHPphob-phil are the contacts of hydrophobic part of ligand with hydrophobic and hydrophilic environment, respectively; MHPphil-phob and MHPphil-phil – the same for hydrophilic part of ligand; HBdcc, HBdcn and HBdnn indicate respectively: charged-charged, charged-neutral and neutral-neutral hydrogen bonds fromed by a ligand donor; HBacc, HBacn and HBann – the same for a ligand acceptor;
**the “goldscore” [24] values were not dissected into fragments and in each case ranking was performed by the integral value of the “goldscore” (see text for discussion);
***the hit-rate 35 was obtained for a combination of fragmental scores for adenine and phosphate, the value 24 was obtained for a whole molecule scoring function (equation not shown since the hit-rate is very poor).
| Score | Hit rates | Relative errors of the weighting coefficients | ||||
|---|---|---|---|---|---|---|
| ADE_score | 30 ± 1 | Constant 0.07 | MHPphob-phob 0.01 | HBdnn 0.02 | Stack 0.36 | Pi_cation 0.23 |
| RIB_score | 12 ± 1 | Constant 0.55 | MHPphob-phob 0.79 | MHPphob-phil 0.98 | Embedding 0.15 | |
| PHOS_score | 23 ± 0 | Constant 0.61 | MHPphil-phil 0.15 | MHPphil-phob 0.21 | Embedding 0.60 | |
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grants 05-04-49283-a, 06-04-49194-a, 07-04-01514-a), by the Russian Federation Federal Agency for Science and Innovations (The State contract 02.467.11.3003 of 20.04.2005, grant “The leading scientific schools” 4728.2006.4).
References
- Gitlin, J.D. Wilson disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar]
- Ferrara, P.; Jacoby, E. Evaluation of the utility of homology models in high throughput docking. J. Mol. Model 2007, 13, 897–905. [Google Scholar]
- Jacobson, K.A.; Kim, S.K.; Costanzi, S.; Gao, Z.G. Purine receptors: GPCR structure and agonist design. Mol. Interv 2004, 4, 337–347. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: current status and future challenges. PROTEINS 2006, 65, 15–26. [Google Scholar]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug. Discov 2004, 3, b935–949. [Google Scholar]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. PROTEINS 2003, 52, 609–623. [Google Scholar]
- Jansen, J.M.; Martin, E.J. Target-biased scoring approaches and expert systems in structure-based virtual screening. Curr. Opin. Chem. Biol 2004, 8, 359–364. [Google Scholar]
- Laederach, A.; Reilly, P.J. Modeling protein recognition of carbohydrates. PROTEINS 2005, 60, 591–597. [Google Scholar]
- Rognan, D.; Lauemoller, S.L.; Holm, A.; Buus, S.; Tschinke, V. Predicting binding affinities of protein ligands from three-dimensional models: application to peptide binding to class I major histocompatibility proteins. J. Med. Chem 1999, 42, 4650–4658. [Google Scholar]
- Pyrkov, T.V.; Kosinsky, Y.A.; Arseniev, A.S.; Priestle, J.P.; Jacoby, E.; Efremov, R.G. Complementarity of hydrophobic properties in ATP-protein binding: a new criterion to rank docking solutions. PROTEINS 2007, 66, 288–298. [Google Scholar]
- Walker, J.; Saraste, M.; Runswick, M.; Gay, N. Distantly related sequences in the α- and β-subunits of ATP-synthase, myosin, kinase and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1982, 1, 945–951. [Google Scholar]
- Traut, T.W. The functions and consensus motifs of 9 types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem 1994, 222, 9–19. [Google Scholar]
- Saraste, M.; Sibbald, P. R.; Wittinghofer, A. The P-loop – a common motif in ATP- and GTPbinding proteins. Trends Biochem. Sci 1990, 15, 430–434. [Google Scholar]
- Babor, M.; Sobolev, V.; Edelman, M. Conserved positions for ribose recognition: importance of water bridging interactions among ATP, ADP and FAD-protein complexes. J. Mol. Biol 2002, 323, 523–532. [Google Scholar]
- Saito, M.; Go, M.; Shirai, T. An empirical approach for detecting nucleotide-binding sites on proteins. Protein. Eng. Des. Sel 2006, 19, 67–75. [Google Scholar]
- Mao, L.; Wang, Y.; Liu, Y.; Hu, X. Molecular determinants for ATP-binding in proteins: a data mining and quantum chemical analysis. J. Mol. Biol 2004, 336, 787–807. [Google Scholar]
- Kuttner, Y.Y.; Sobolev, V.; Raskind, A.; Edelman, M. A consensus-binding structure for adenine at the atomic level permits searching for the ligand site in a wide spectrum of adenine-containing complexes. PROTEINS 2003, 52, 400–411. [Google Scholar]
- Cappello, V.; Tramontano, A.; Koch, U. Classification of proteins based on the properties of the ligand-binding site: the case of adenine-binding proteins. PROTEINS 2002, 47, 106–115. [Google Scholar]
- Denissiouk, K.A.; Rantanen, V.V.; Johnson, M.S. Adenine recognition: a motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins. PROTEINS 2001, 44, 282–291. [Google Scholar]
- Khedkar, S.A.; Malde, A.K.; Coutinho, E.C.; Srivastava, S. Pharmacophore modeling in drug discovery and development: an overview. Med. Chem 2007, 3, 187–197. [Google Scholar]
- Chang, C.; Ekins, S.; Bahadduri, P.; Swaan, P. W. Pharmacophore-based discovery of ligands for drug transporters. Adv. Drug. Deliv. Rev 2006, 58, 1431–1450. [Google Scholar]
- Efremov, R.G.; Chugunov, A.O.; Pyrkov, T.V.; Priestle, J.P.; Arseniev, A.S.; Jacoby, E. Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem 2007, 14, 393–415. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic. Acid. Res 2000, 28, 235–242. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R.D. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of ALOGP and CLOGP methods. J. Phys. Chem 1998, 102, 3762–3772. [Google Scholar]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C; Mainz, D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49, 6177–6196. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Pyrkov, T.V.; Efremov, R.G. A Fragment-Based Scoring Function to Re-rank ATP Docking Results. Int. J. Mol. Sci. 2007, 8, 1083-1094. https://doi.org/10.3390/i8111083
AMA Style
Pyrkov TV, Efremov RG. A Fragment-Based Scoring Function to Re-rank ATP Docking Results. International Journal of Molecular Sciences. 2007; 8(11):1083-1094. https://doi.org/10.3390/i8111083
Chicago/Turabian StylePyrkov, Timothy V., and Roman G. Efremov. 2007. "A Fragment-Based Scoring Function to Re-rank ATP Docking Results" International Journal of Molecular Sciences 8, no. 11: 1083-1094. https://doi.org/10.3390/i8111083