The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells

Department of Bioscience Technology and Center for Nanotechnology, Chung Yuan Christian University, Chung Li, Taiwan 32023
Int. J. Mol. Sci. 2007, 8(11), 1177-1195; https://doi.org/10.3390/i8111177
Submission received: 12 November 2007
/
Revised: 16 November 2007
/
Accepted: 21 November 2007
/
Published: 27 November 2007
(This article belongs to the Special Issue Natural Compounds for Cancer Treatment and Prevention)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Ginkgolide B, the major active component of Ginkgo biloba extracts, can both stimulate and inhibit apoptotic signaling. Here, we demonstrate that ginkgolide B can induce the production of reactive oxygen species in MCF-7 breast cancer cells, leading to an increase in the intracellular concentrations of cytoplasmic free Ca2+ and nitric oxide (NO), loss of mitochondrial membrane potential (MMP), activation of caspase-9 and -3, and increase the mRNA expression levels of p53 and p21, which are known to be involved in apoptotic signaling. In addition, prevention of ROS generation by pretreatment with N-acetyl cysteine (NAC) could effectively block intracellular Ca2+ concentrations increases and apoptosis in ginkgolide B-treated MCF-7 cells. Moreover, pretreatment with nitric oxide (NO) scavengers could inhibit ginkgolide B-induced MMP change and sequent apoptotic processes. Overall, our results signify that both ROS and NO played important roles in ginkgolide B-induced apoptosis of MCF-7 cells. Based on these study results, we propose a model for ginkgolide B-induced cell apoptosis signaling cascades in MCF-7 cells.
1. Introduction
Ginkgolide B, the major component of extracts from leaves of Ginkgo biloba L. used in traditional Chinese herbal medicine, exerts a wide range of biological activities, including antiangiogenesis and antioxidation [1–3]. Ginkgolides are terpenoid compounds, and ameliorate tissue irrigation by augmenting cellular energy in the arterial, venous, and capillary circulation. In particular, ginkgolide B possesses antioxidant activity, and is a potent peroxy radical scavenger [4]. Ginkgolides decrease singlet oxygen, hydrogen peroxide and hydroxyl radical production, supporting their role as antioxidants [5]. Recent reports show that the increase in free radicals and associated lipid peroxidation reactions are suppressed in the central nervous system and heart upon treatment with extracts of Ginkgo biloba leaves [6,7]. In addition, Ginkgo biloba extracts inhibit β-amyloid aggregation-induced caspase-3 activation and cell apoptosis [8]. Another study demonstrated that Ginkgo biloba extracts induce injury of oral cavity tumors via apoptosis processes [9]. Thus, extracts of this compound appear to have both anti- and pro-apoptotic effects. However, while multiple biological functions of Ginkgo biloba extracts have been identified to date, the precise molecular mechanisms underlying these actions remain to be established.
Apoptosis plays an important role in the embryogenesis and homeostasis of multi-cellular organisms, and impairment of apoptotic function has been associated with several human diseases, including neurodegenerative disorders and cancer [10]. Numerous chemical and physical treatments capable of inducing apoptosis can stimulate oxidative stress via generation of ROS in cells [11–13], suggesting that there is a close relationship between oxidative stress and apoptosis. Although the precise molecular mechanisms underlying apoptosis have not been clearly defined, a number of cysteine proteases called caspases are thought to play important roles [14,15], as do members of the Bcl-2 family [16], which regulate mitochondrial membrane potential changes and the release of mitochondrial cytochrome c by modulating the permeability of the outer mitochondrial membrane. Nitric oxide (NO) is an important second messenger that is involved in a variety cellular responses and biological functions, including tumor development, metastasis and apoptosis [17–19]. Recent studies have demonstrated that NO is largely produced in mitochondria, through the actions of a Ca2+-sensitive mitochondrial NO synthase (NOS) [20,21]. This NOS-mediated NO production could control oxygen consumption and mitochondrial membrane potential through cytochrome c oxidase; the NO molecule could then be reactivated with superoxide to produce peroxynitrite, which could further modify its target substrates and induce oxidative stress [22–24]. Several recent studies have shown that oxidative stress and Ca2+ influxes act as upstream regulators for mitochondrial NOS activity [25,26]. However, the biochemical events involved in ginkgolide B-induced cell injury and the regulatory mechanisms underlying these effects are still unclear.
To elucidate the precise regulatory mechanisms of ginkgolide B-triggered cytotoxicity, we examined the effects of ginkgolide B on human breast cancer MCF-7 cells. Our results revealed that ginkgolide B treatment triggers apoptotic biochemical changes such as oxidative stress generation, Ca2+ influx, NO production, loss of MMP, increased P53 and P21 expression levels, and caspase-9/-3 activation in MCF-7 cells.
2. Results and Discussion
MCF-7 cells were incubated with various doses of ginkgolide B, and cell viability was determined. Our results revealed that cell viability decreases in response to dose-dependent treatment of 5 to 80 μM ginkgolide B (Figure 1A). To examine the cell death mode underlying this decreased viability, cells apoptosis and necrosis were measured by propidium iodide/Hoechst 33342 staining and TUNEL assay. Our results revealed that ginkgolide B increased the apoptosis-associated parameter in a dose-dependent manner (Figure 1B and 1C). In addition, we tested the effect of vehicle alone (0.5% DMSO) in each experiment. Our results showed that treatment with vehicle alone had no effect on cell apoptosis (data not shown).
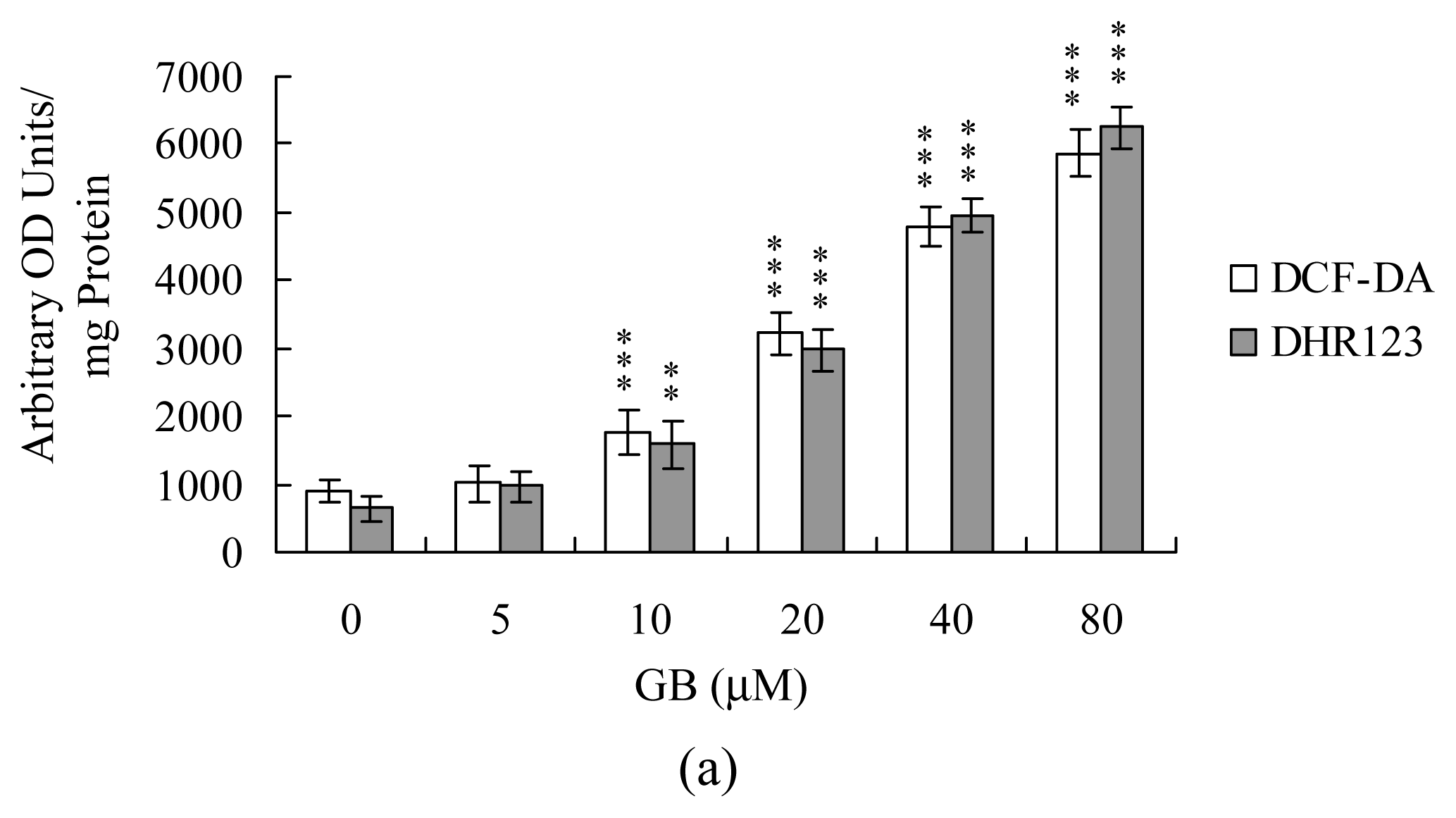
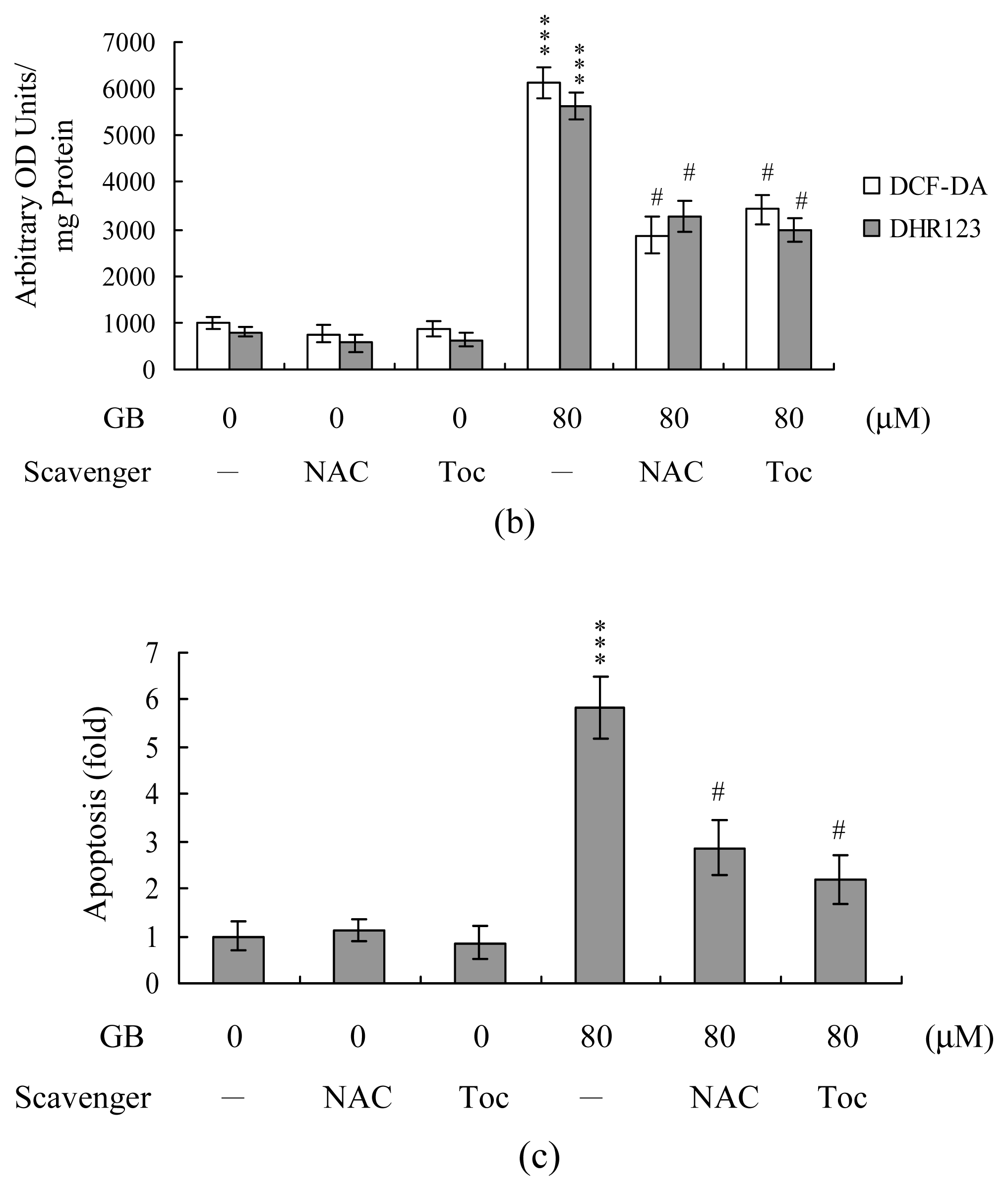
Since our previous studies demonstrated that numerous stimuli can trigger cell apoptosis via ROS generation [27, 28], we used DCF-DA and dihydrorhodamine 123 (DHR 123) to examine whether ROS formation occurred in MCF-7 cells treated with ginkgolide B. Our results showed that ginkgolide B (20–80 μM) treatment stimulated ROS generation about 4–10-fold compared to the untreated control group (Figure 2A). Importantly, pretreatment with two commonly-used ROS scavengers, N-acetyl cysteine (NAC) and α-tocopherol, effectively prevented ROS generation and apoptosis triggered by ginkgolide B (Figure 2B and 2C). These results suggest that ROS generation plays an important regulator in ginkgolide B-induced apoptosis.
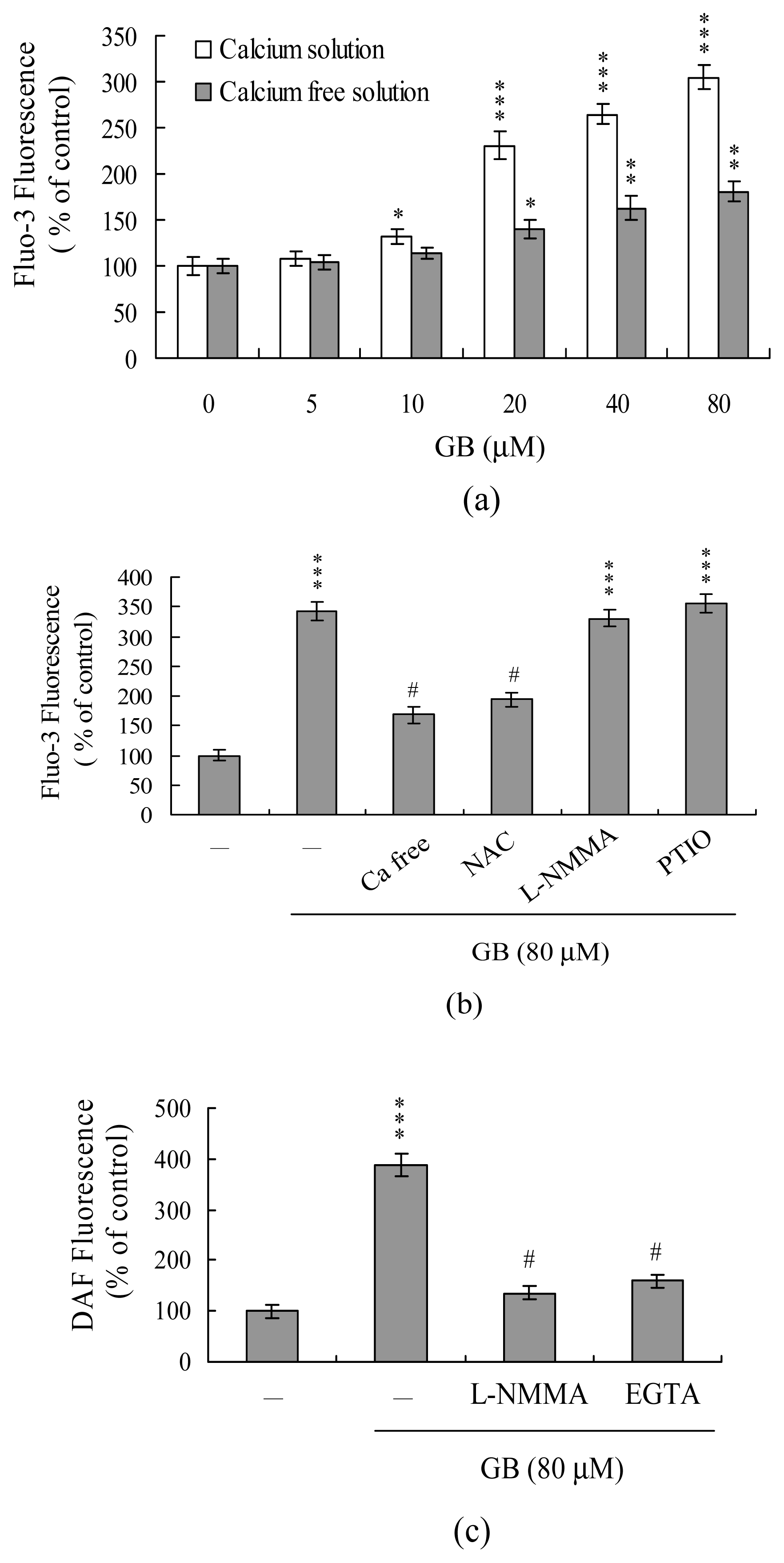
Changes in intracellular Ca2+ concentration ([Ca2+]i) in ginkgolide B-treated MCF-7 were detected using the Fluo-3AM fluorescence dye. We found that treatment with ginkgolide B elicited an increase in [Ca2+]i (Figure 3A). Furthermore, cells cultured in Ca2+-containing medium showed a ~3-fold increase of [Ca2+]i following treatment with 80 μM ginkgolide B, whereas this increase was largely, but not completely, blocked in treated cells cultured in Ca2+-free cultured medium (Figure 3A). These findings indicate that the rise in [Ca2+]i could be primarily attributed to an influx of external Ca2+, with a smaller portion due to calcium release from intracellular stores, such as those found in the endoplasmic reticulum, mitochondria, nucleus and/or calcium-binding proteins (Figure 3A). Our results further revealed that PTIO, an inhibitor of NOS and scavenger of NO, and L-NMMA, an inhibitor of NOS, had little effect on ginkgolide B-induced [Ca2+]i increases, whereas pretreatment with NAC significantly decreased this effect (Figure 3B). These results suggest that the elevation of [Ca2+]i induced by ginkgolide B treatment may be regulated by ROS generation but not by NO. We further used the NO-sensitive dye, DAF-2DA, to measure intracellular NO generation during ginkgolide B-induced cell apoptosis. Our results revealed that intracellular NO levels increased in MCF-7 cells treated with ginkgolide B (Figure 3C). However, this increase could be prevented by pretreatment of cells with the NOS inhibitor, L-NMMA (Figure 3C), or 400 μM EGTA (a Ca2+ chelator) (Figure 3C). These results suggest that intracellular Ca2+ levels play an important role in the NOS activation and NO increases observed in MCF-7 cells treated with ginkgolide B.
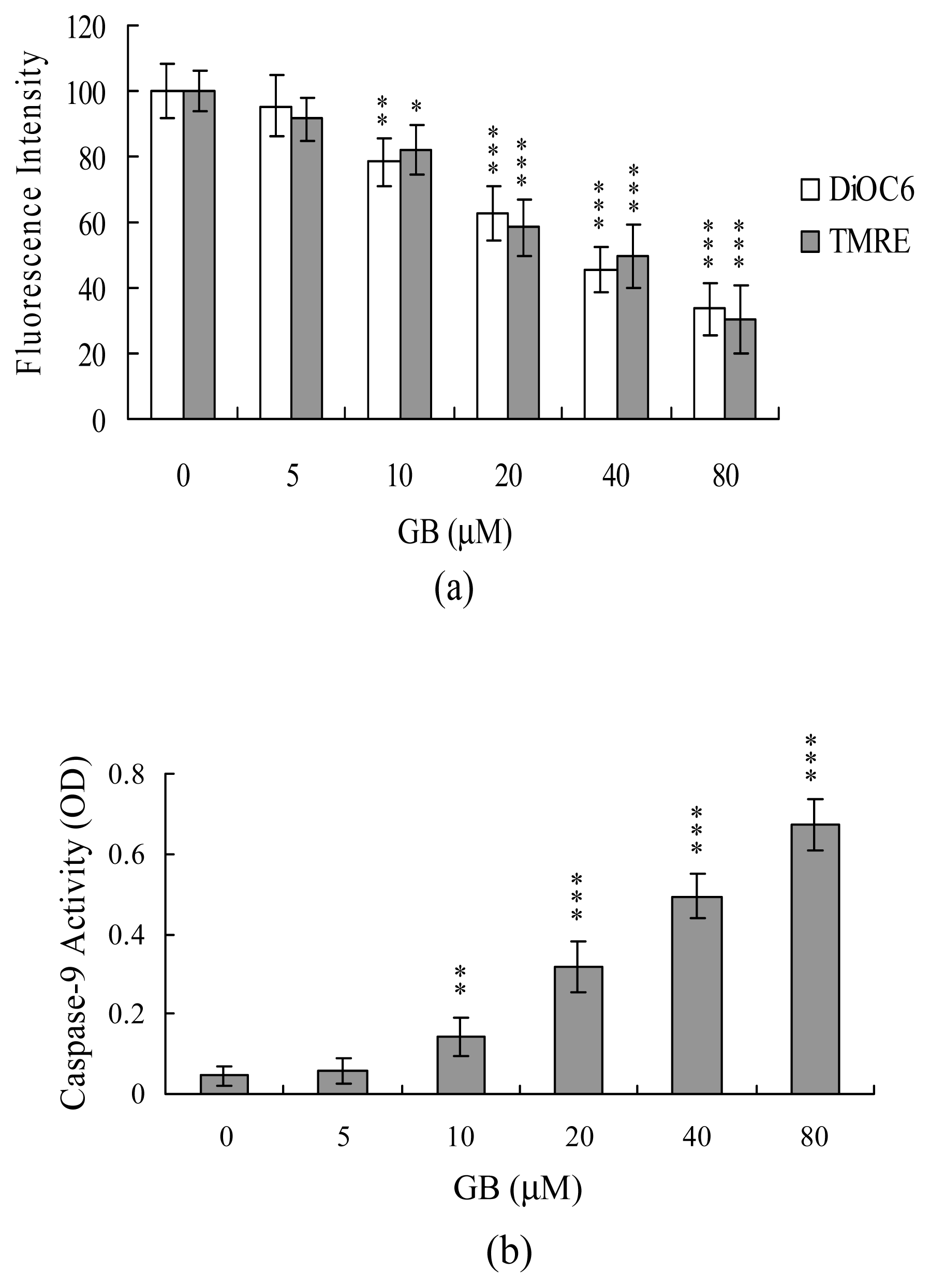
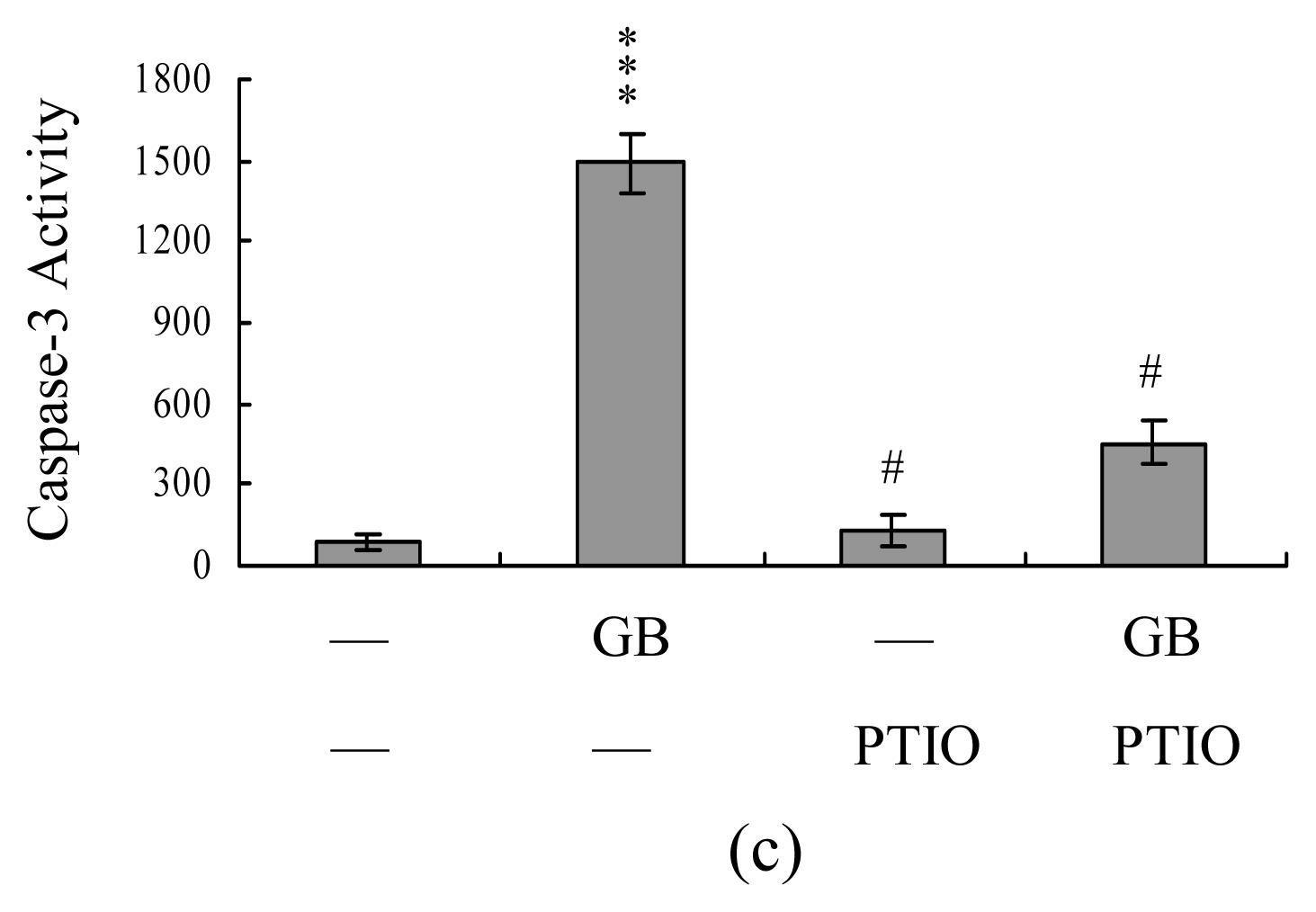
We next analyzed MMP change, a major apoptotic event during mitochondrial-mediated apoptotic processes [29]. Treatment of MCF-7 cells with ginkgolide B decreased DiOC6(3) and TMRE uptake into mitochondria, indicating that ginkgolide B induces significant loss of MMP (Figure 4A). In addition, we monitored the activations of caspase-9 and caspase-3, which are known to be involved in mitochondria-dependent apoptotic pathway, and found that treatment of MCF-7 cells with 5–80 μM ginkgolide B stimulated the activation of caspase-9 (Figure 4B) and caspase-3 (Figure 4C). Importantly, both MMP change and caspases activation were significantly inhibited by incubation of cells with 20 μM PTIO prior to treatment with 80 μM ginkgolide B (Figure 5A–5C). These results indicate that increases in NO level may act as an upstream regulator for MMP change and activation of caspase-9 and -3 during the ginkgolide B-induced apoptosis of MCF-7 cells.
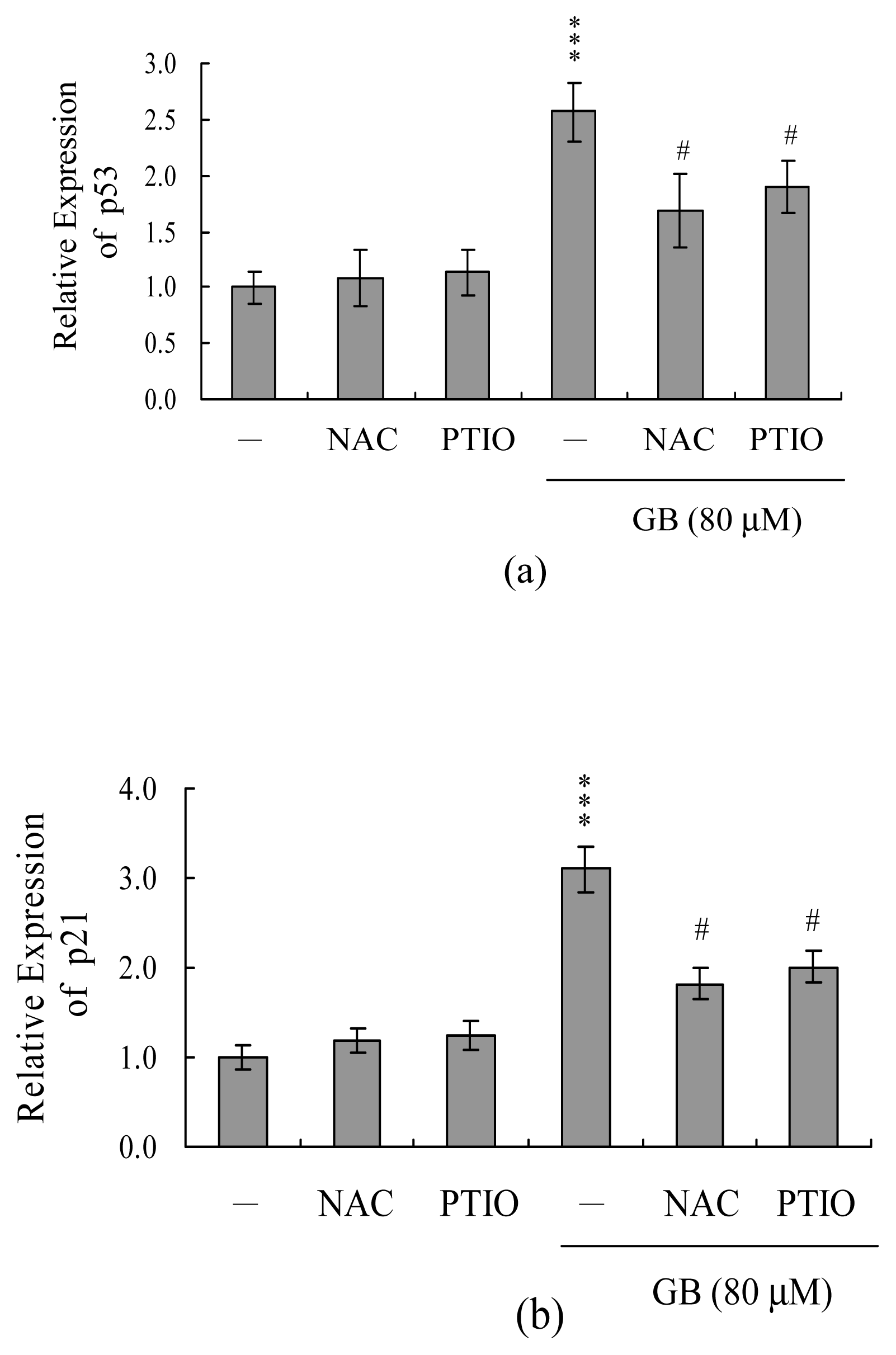
The results from our real-time RT-PCR analyses showed that p53 and p21 were significantly up-regulated at the mRNA levels in MCF-7 cells treated with ginkgolide B, but these changes could be blocked by pretreatment with NAC or PTIO (Figure 6A and 6B).
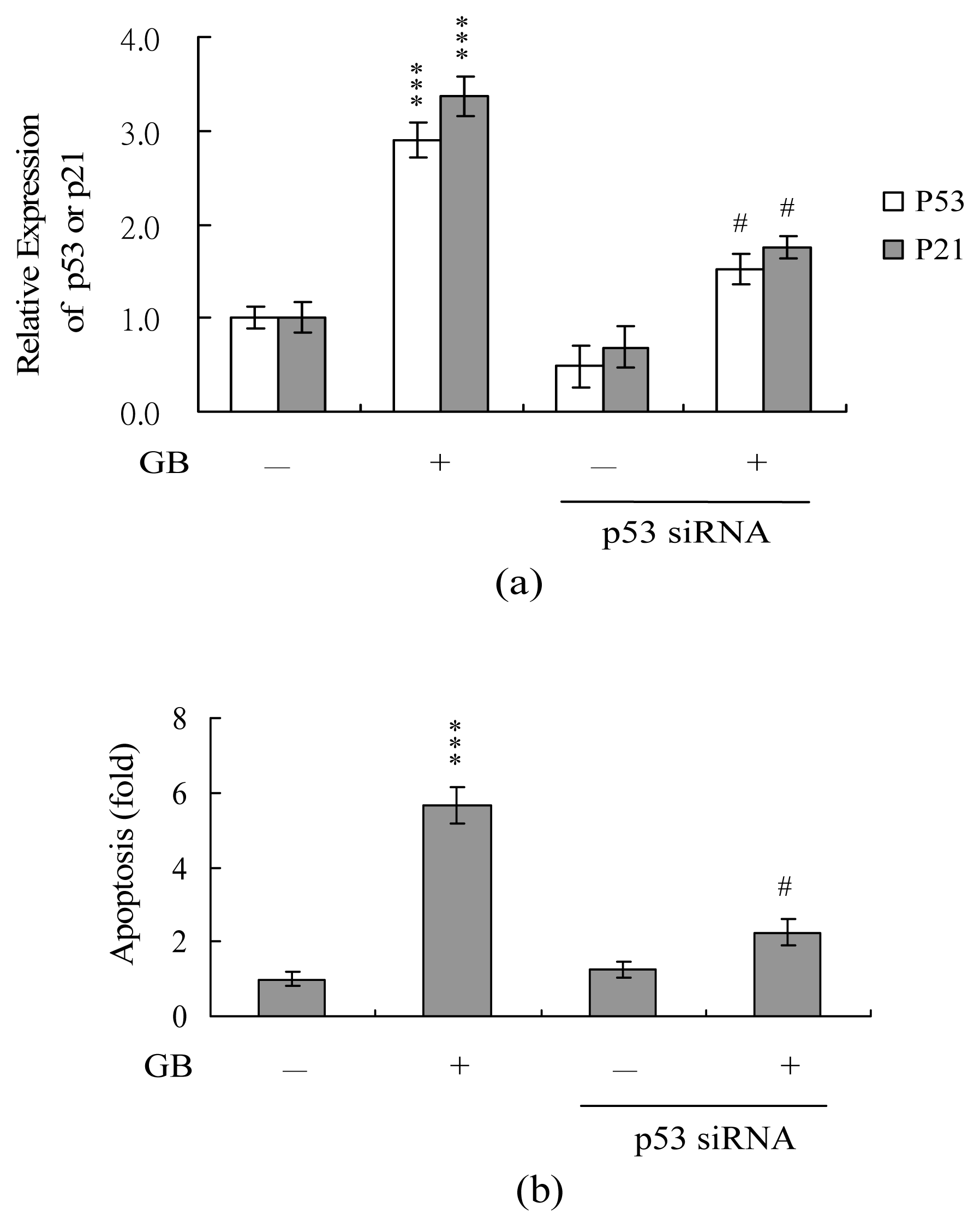
To further determine the role of p53 in ginkgolide B-induced apoptosis, we used targeted siRNAs to decrease the expression levels of p53 in MCF-7 cells, and then incubated the cells with ginkgolide B for 24 h and tested for cell viability. The siRNA knockdown significantly decreased the protein expression levels (data not shown) and mRNA levels of p53 and p21 (Figure 7A) in ginkgolide B-treated MCF-7 cells, and this decrease in p53 expression level was associated with a significant decrease in ginkgolide B-induced apoptosis (Figure 7B). These results suggest that ginkgolide B treatment up-regulates p53 in MCF-7 cells, and this effect is involved in subsequent apoptosis of the treated cells.
Previous reports showed that Ginkgo biloba extract and its derivative components can induce apoptosis and inhibit cell proliferation in various cell types, including oral cavity cancer, human breast cancer, colorectal cancer, and hepatoma cells [9, 30, 31]. We previously also showed that ginkgolides could trigger cell death and decreased cell proliferation in blastocysts through ROS generation and JNK activation [12, 32]. Oxidative stress is a stimulator of cell responses such as apoptosis [33, 34]. An earlier study reports that ginkgolide B inhibits ROS generation and apoptosis via its antioxidant properties in β-amyloid-induced apoptosis [8].
Importantly, ginkgolide B directly induces oxidative stress generation in MCF-7 in our experiments (Figure 2). In addition, pretreatment with anti-oxidants effectively prevents ROS-induced apoptotic biochemical changes (Figure 2 and 3). Several reports demonstrate that ROS is an important upstream regulator of caspase activation during UV-, methylglyoxal-, and osmotic shock-induced apoptosis in various mammalian cells [27, 28, 35]. Since recent papers have shown that the addition of some compounds to commonly-utilized cell culture media can trigger generation of ROS such as hydrogen peroxide [36, 37], we incubated ginkgolide B and culture medium together, and measured ROS by the ferrous iron oxidation-xylenol orange method [36]. The results (data not shown) showed no detectable artifactual ROS generation under these conditions. These results, coupled with our findings, strongly imply that oxidative injury plays a pivotal role in ginkgolide B-induced apoptosis. However, the underlying mechanism of ginkgolide B-triggered ROS formation remains to be investigated.
Mitochondria act as important conduits for signals during programmed cell death, and loss of mitochondrial integrity is promoted or inhibited by many key regulators of apoptosis [38, 39]. For instance, various cellular stress conditions, including heat shock, DNA damage and oxidative stress, result in caspase activation through loss of MMP and cytochrome c release from the mitochondrial intermembrane space into the cytoplasm [39, 40]. To further clarify the precise mechanisms underlying ginkgolide B-induced apoptosis, we examined MMP changes. Ginkgolide B induced loss of MMP in a dose-dependent manner (Figure 4A). In addition, our study also found that PTIO pretreatment caused ~70% reduction in ginkgolide B-induced NO increases (data not shown). This study result explains how ginkgolide B (80 μM) could decrease DiOC6(3) and TMRE uptake into mitochondria by 65%. However, pretreatment with PITO only reversed by 30% the inhibitory effect of ginkgolide B on DiOC6(3) and TMRE uptake. Moreover, caspases-9 and -3 were activated by ginkgolide B (Figure 4B and 4C). Inhibition of NO generation by PTIO led to a decrease in ginkgolide B-triggered mitochondria-dependent apoptotic changes (Figure 5). We propose that NO is an important regulator of ginkgolide B-induced mitochondria-dependent apoptotic processes.
Previous studies have shown that increases in intracellular calcium levels can play an important role in regulating cell death [20, 41, 42]. In this study, we elucidated whether ginkgolide B treatment induced cell apoptosis through intracellular calcium increases. We observed increases in [Ca2+]i following ginkgolide B treatment, and found that this increase in [Ca2+]i was largely due to an influx of Ca2+ from the extracellular medium (Figure 3A). This effect could be significantly blocked by NAC (Figure 4B), indicating that ginkgolide B-induced ROS generation is responsible for the elevation of intracellular calcium concentrations in ginkgolide B-treated MCF-7 cells.
NO is a product produced endogenously from NADPH, O2 and L-arginine catalyzed by nitric oxide synthase. A recent study demonstrated that NO production was involved in apoptosis triggered by several different types of stimuli [20, 21]. The regulatory actions of NO on the mitochondrial apoptotic signaling pathways are well documented. Decreased ratios of Bcl-2/Bax and inhibition of the electron transport are regulatory mechanisms of NO-mediated apoptosis [43]. Decrease on Bcl-2/Bax ratios and impairment of mitochondrial electron transport may cause release of cytochrome c which involved in the control of cell apoptosis. In addition, tamoxifen treatment increased intramitochondrial Ca2+ concentrations, leading to stimulation of mitochondrial NO synthase activity and enhancement of NO production in rat livers and human breast cancer MCF-7 cells [21]. Here, we observed NO generation following ginkgolide B treatment of MCF-7 cells, with intracellular NO levels ~4-fold higher in treated cells versus untreated controls (Figure 3C). Pretreatment with EGTA in the extracellular medium significantly prevented this increase in intracellular NO (Figure 3C), indicating that NO production in ginkgolide B-treated MCF-7 cells is dependent on the intracellular calcium concentration. The regulatory role of NO in cell apoptosis is very complex, and studies have shown that NO-mediated apoptotic effects are modulated by different mechanisms in different cell types [19, 44]. A previous study showed that NOS substrates or NO donors could inhibit photodynamic treatment-induced apoptosis in CCRF-CEM cells [45]. Here, we found that PTIO treatment attenuated loss of MMP and decreased caspases activation (Figure 5), suggesting that NO generation may be an important mediator of apoptosis in ginkgolide B-treated MCF-7 cells.
It is well known that NO-mediated apoptotic processes are associated with p53 gene activation, which is essential for regulation of cell cycle and/or apoptotic signaling occurring through p21Waf1/Cip1 or Bax [46, 47]. In this study, we found that both p53 and p21 mRNA levels were up-regulated by treatment with ginkgolide B, and this up-regulation was blocked by pretreatment with PTIO (Figures 6A and 6B). Furthermore, siRNA-mediated knockdown of p53 mRNA and protein expression levels prevented ginkgolide B-induced increases in the mRNA and protein levels of p21, and decreased subsequent apoptosis (Figures 7A and 7B), whereas p21 knockdown blocked ginkgolide B-triggered apoptosis in MCF-7 cells, but had no effect on the expression of p53 (data not shown). Our previous study has demonstrated that p53 gene activation is an upstream regulator for p21 gene expression [48]. These results indicate that the genes encoding p53 and p21 are activated during ginkgolide B-induced apoptosis of MCF-7 cells, and further suggest that NO plays an important role in regulating gene expression and cell death in ginkgolide B-treated MCF-7 cells. Moreover, comparison of cell groups in pre-treatment with NAC, PTIO, or NAC/PTIO and then incubated with ginkgolide B (80 μM) for 24 h, the study showed that pre-treatment of cells with NAC, PTIO, or NAC/PTIO had no significant difference effects on prevention of ginkgolide B-triggered up-regulation of p53 and p21 mRNA expression levels. These observations taken together with the findings that ROS generation and NO increases triggered by ginkgolide B can be blocked by NAC, support the hypothesis that ROS generation is an upstream regulator of NO increases and sequent apoptotic biochemical changes in ginkgolide B-induced apoptotic processes.
The studies have shown that ginkgolide B can both stimulate and inhibit apoptotic signaling. Our previous study revealed that 5–10 μM ginkgolide B treatment of embryonic stem (ESC-B5) cells induced apoptosis via ROS generation, JNK activation, loss of mitochondrial membrane potential and activation of caspase-3 [12]. In addition, our study also demonstrate that 5–10 μM ginkgolide B treatment of mouse blastocysts induces apoptosis, decreases cell numbers, retards early postimplantation blastocyst development, and increases early-stage blastocyst death [32]. In contrast, amyloid-β1–42-induced apoptosis in neurons were inhibited by ginkgolide B (10 nM-10 μM for 3h) [3]. Importantly, our previous study found that ethanol (50–400 mM) induced apoptotic cell death in human Hep G2 cells, and that this effect was inhibited by low (5–25 μM) doses of ginkgolide B, but enhanced by high (50–100 μM) doses of ginkgolide B [49]. The result point out that ginkgolide B is a ROS scavenger or enhancer for ethanol-derived free radical generation, and that this molecule has the ability to inhibit or promote cell death depending on the treatment dosage [49]. In the present study, we show that ginkgolide B at doses higher than 10 μM induces ROS generation and sequent apoptotic biochemical changes in MCF-7 cells (Figures 1–6). Taken together these observations and our research results seem to implicate that there is some cell type specificity and treatment protocol (i.e., treatment period and dosage of ginkgolide B) may determine the effect of ginkgolide B. However, the molecular mechanisms of ginkgolide B on cell apoptosis need further investigation for the regimen employed.
3. Conclusions
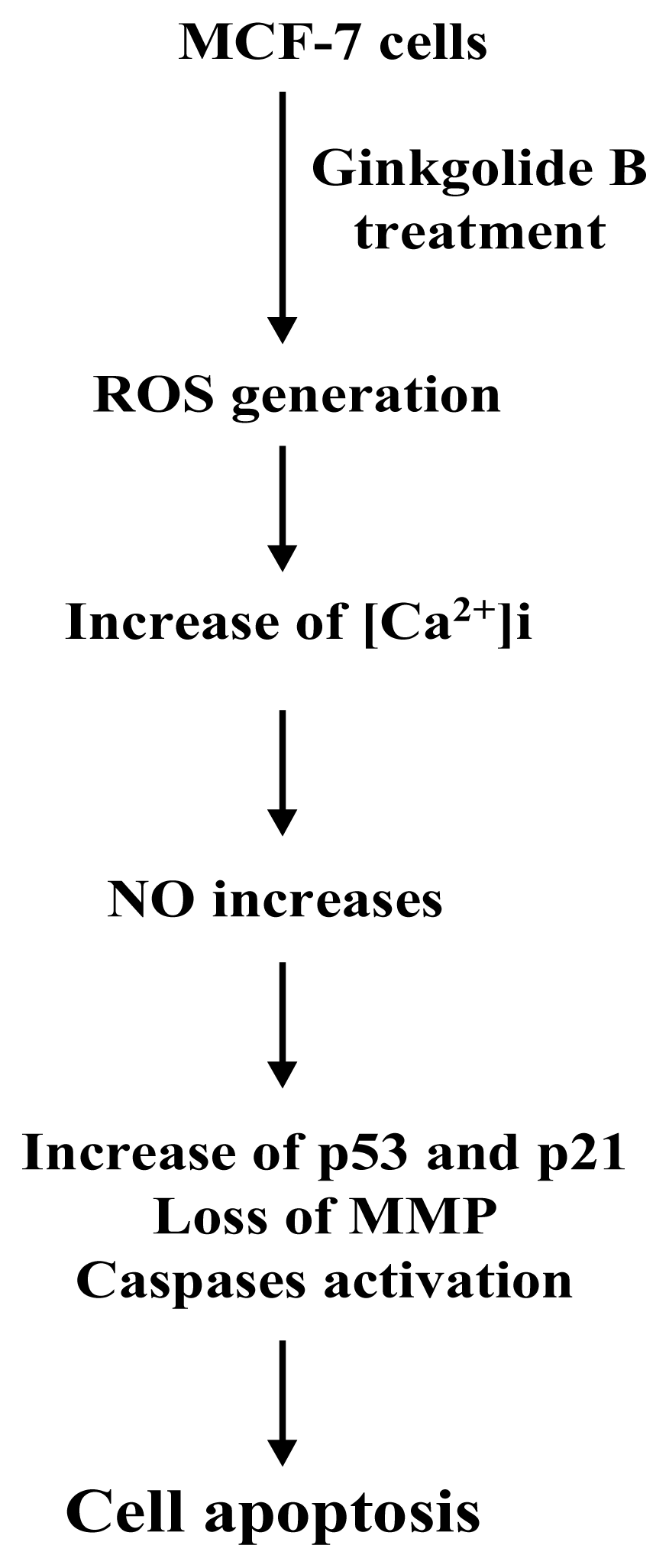
Based on the results of the present study, we propose a possible model of ginkgolide B-induced cell apoptosis signaling in MCF-7 cells (Figure 8). Our results collectively show that both intracellular ROS and NO play critical roles in ginkgolide B-induced cell apoptosis of MCF-7 cells.
4. Experimental Section
4.1. Chemicals
Ginkgolide B, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), N-acetyl cysteine (NAC), α-tocopherol, 2′,7′-dichlorofluorescein diacetate (DCF-DA), propidium iodide, Hoechst 33342, ethyleneglycol-bis(β-aminoethylether)tetraacetic acid (EGTA) and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Sigma (St. Louis, MO). Z-DEVD-AFC was obtained from Calbiochem (La Jolla, CA).
4.2. Cell culture and ginkgolide B treatment
Human breast cancer MCF-7 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells (~5–6 × 106) were plated on 60 mm culture dishes and incubated in media containing various concentrations of ginkgolide B for 24 h. The cells were then washed twice with ice-cold PBS and lysed in lysis solution (600 μL, 20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mM benzamidine, 1 mM phenyl-methylsulfonyl fluoride, 50 mM NaF, 20 mM sodium pyrophosphate and 1 mM sodium orthovanadate) on ice for 10 min. The cell lysates were collected, sonicated on ice for 3 × 10 sec, and then centrifuged at 15,000 × g for 20 min at 4°C. The resulting supernatants were used as the cell extracts.
4.3. MTT assay
The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) test is a colorimetric assay that measures the percentage of cell survival. Following treatment of cells with ginkgolide B, 100 μl of 0.45 g/l MTT solution was added to each well in 96 wells culture plate. Cells were incubated at 37°C for 60 min to allow color development, and 100 μl of 20% SDS in DMF-H2O (1:1) solution was added to each well to halt the reaction. The plates were then incubated overnight at 37 °C to dissolve the formazan products. The results were analyzed by spectrophotometry using an ELISA reader at a wavelength of 570 nm.
4.4. Assessment of necrosis and apoptosis
Oligonucleosomal DNA fragmentation (a hallmark of apoptosis) was measured using the Cell Death Detection ELISAplus kit (Roche Molecular Biochemicals, Mannheim, Germany). Cells (1×105) were treated with or without the indicated concentrations of ginkgolide B at 37 °C for 24 h, the procedures were performed according to the manufacturer’s protocol, and spectrophotometric data were obtained using an ELISA reader at 405 nm. In addition, cells were incubated with propidium iodide (1 μg/mL) and Hoechst 33342 (2 μg/mL) at room temperature for 10 min, and fluorescent microscopy was used to identify the percentage of propidium iodide-impermeable cells having condensed/fragmented nuclei (apoptotic) and the percentage of propidium iodide-permeable cells (necrotic). In each experiment, 7–10 independent fields (~600–1000 nuclei in total) were counted per condition.
4.5. ROS assay
ROS arbitrary units were measured using 2′-7′-dichlorofluorescein diacetate (DCF-DA) or dihydrorhodamine 123 (DHR 123) dye. Cells (1.0 × 106) were incubated in 50 μL PBS containing 20 μM DCF-DA or 20 μM DHR 123 for 1 h at 37°C, and relative ROS units were determined using a fluorescence ELISA reader (excitation 485 nm, emission 530 nm). An aliquot of the cell suspension was lysed, the protein concentration was determined, and the results were expressed as arbitrary absorbance units/mg protein.
4.6. Detection of mitochondrial membrane potential (MMP)
Cells were dispensed into 96-well plates, grown for 24 h, and incubated with various concentrations of ginkgolide B for 24 h. Fluorescent dyes, DiOC6(3) (40 nM) or TMRE (1 μM), were added to each well, incubated for 15 min, and fluorescence measured with a plate spectrofluorometer [excitation: 485 nm (DiOC6(3)) or 535 nm (TMRE); emission: 535 nm (DiOC6(3)) or 590 nm (TMRE)] [50, 51].
4.7. Caspase activity assays
4.8. Detection of intracellular calcium concentration ([Ca2+]i )
The [Ca2+]i was detected with Fluo-3 AM fluorescence dye, using a modification of the previously reported method [20, 53]. Briefly, Cells were treated with ginkgolide B, harvested and washed, and then loaded with 6 μM Fluo-3 AM in standard medium (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5.6 mM glucose, 1.5 mM CaCl2, and 20 mM Hepes, pH of 7.4). After 30 min, the cells were washed three times with PBS and then resuspended in standard medium or Ca2+-free standard medium. The fluorescence intensity of Fluo-3 was determined using a fluorescence spectrophotometer (Hitachi, F-2000; excitation at 490 nm, emission at 526 nm).
4.9. Detection of intracellular NO content
The DAF-2DA fluorescence dye was used to detect intracellular NO, according to a modification of the previously reported method [20, 54]. Briefly, treated or control cells were collected and washed, and then incubated with 3 μM DAF-2DA. After 60 min, the cells were washed three times with PBS and the fluorescence intensity was measured by a fluorescence spectrophotometer (Hitachi, F-2000; excitation at 485 nm, emission at 515 nm).
4.10. Real-time RT-PCR assay
Total RNA was extracted with the TRIzol reagent (Life Technologies) and purified with an RNeasy Mini kit (Qiagen), according to the manufacturers’ protocols. Real-time PCR was carried out with an ABI 7000 Prism Sequence Detection System (Applied Biosystems). The β-actin mRNA levels were quantified as an endogenous control, and used for normalization. The primers used for PCR were as follows: p53, 5′-CCC ATC CTC ACC ATC ATC AC-3′ and 5′-GTC AGT GGG GAA CAA GAA GTG-3′; p21, 5′-GCC GAA GTC AGT TCC TTG TGG A-3′ and 5′-GTG GGC GGA TTA GGG CTT-3′.
4.11. siRNA knockdown
Lipofectamine was used to transfect MCF-7 cells with 150 nM of siRNA for targeting against p53 (5′-GACUCCAGUGGUAAUCUACTT-3′; sip53) or a scrambled control duplex (5′-GCGCGCUUUGUAGGAUUCG-3′; siScr). Twenty-four hours post-transfection, fresh culture medium was added, and the cells were treated with or without ginkgolide B for another 24 h.
4.12. Statistics
Data were analyzed using one-way ANOVA, and the differences were evaluated using a Student’s t-test and analysis of variance. A P-value <0.05 was considered significant.
Figure 1.
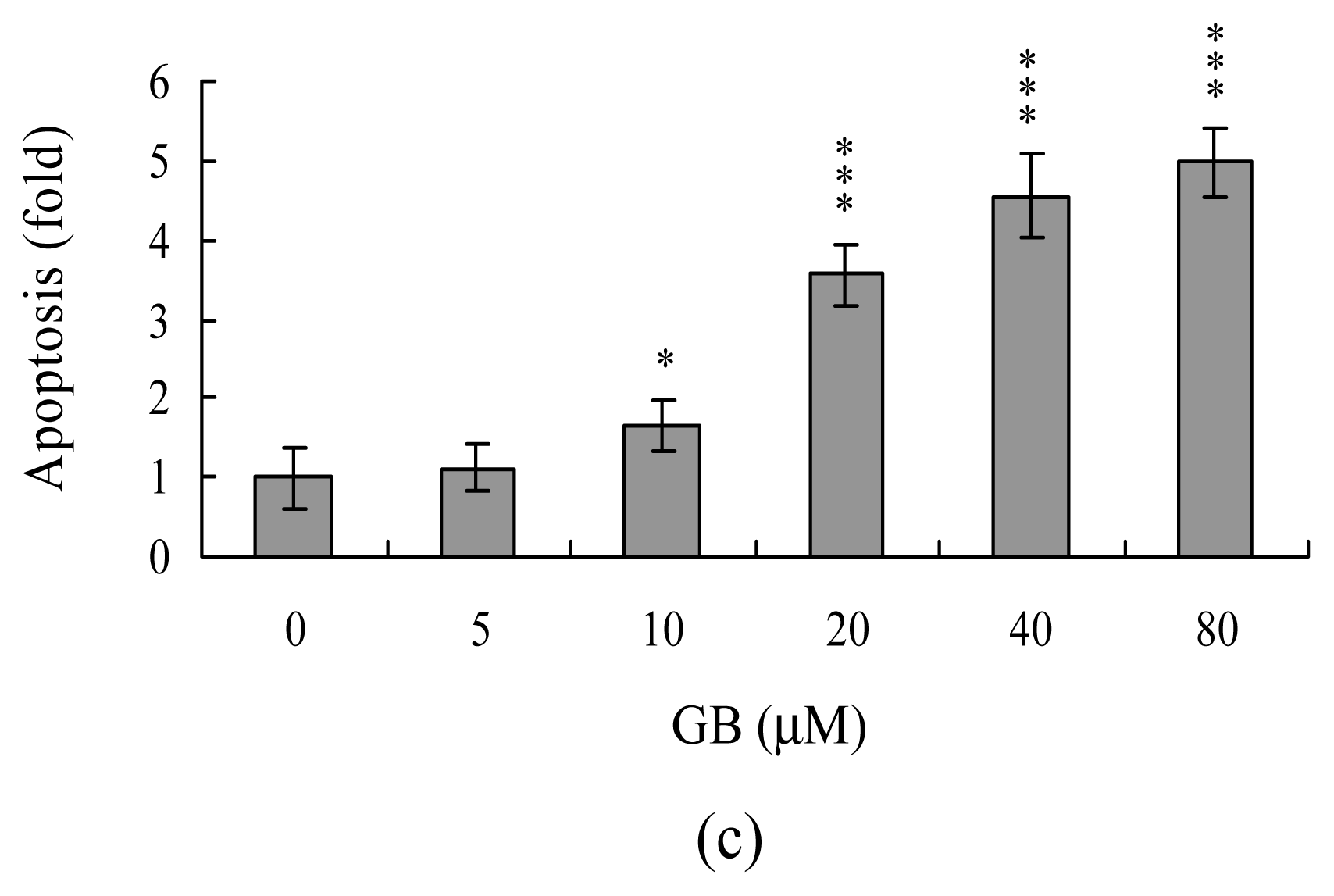
Effects of ginkgolide B on MCF-7 cells. Cells were incubated with or without various concentrations of ginkgolide B (GB) for 24 h. (A) Cell viability was determined by MTT assay; (B) The percentages of apoptosis and necrosis were determined by propidium iodide (PI) and Hoechst 33342 staining; (C) Apoptosis was evaluated using the Cell Death Detection ELISA kit. Values are presented as means ± SD of six determinations. *P<0.05, **P<0.01 and ***P<0.001 versus the control (ginkgolide B-free) group.
Figure 1.
Effects of ginkgolide B on MCF-7 cells. Cells were incubated with or without various concentrations of ginkgolide B (GB) for 24 h. (A) Cell viability was determined by MTT assay; (B) The percentages of apoptosis and necrosis were determined by propidium iodide (PI) and Hoechst 33342 staining; (C) Apoptosis was evaluated using the Cell Death Detection ELISA kit. Values are presented as means ± SD of six determinations. *P<0.05, **P<0.01 and ***P<0.001 versus the control (ginkgolide B-free) group.


Figure 2.
Ginkgolide B induces ROS generation in MCF-7 cells. Cells were incubated with 20 μM DCF-DA or 20 μM dihydrorhodamine 123 (DHR 123) for 1 h, and treated with various concentrations of ginkgolide B (GB) for another 24 h. (A) ROS generation was assayed by DCF-DA or DHR 123 fluorescence, and expressed as absorbance/mg of protein; (B, C) Cells were pre-incubated with N-acetyl cysteine (NAC; 300 μM) or α–tocopherol (Toc; 300 μM) for 30 min and then treated with ginkgolide B (80 μM) for another 24 h. ROS generation was assayed using DCF-DA or DHR 123 dye (B). Apoptosis was evaluated using the Cell Death Detection ELISA kit (C). The data are representative of eight independent experiments. **P<0.01 and ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.
Figure 2.
Ginkgolide B induces ROS generation in MCF-7 cells. Cells were incubated with 20 μM DCF-DA or 20 μM dihydrorhodamine 123 (DHR 123) for 1 h, and treated with various concentrations of ginkgolide B (GB) for another 24 h. (A) ROS generation was assayed by DCF-DA or DHR 123 fluorescence, and expressed as absorbance/mg of protein; (B, C) Cells were pre-incubated with N-acetyl cysteine (NAC; 300 μM) or α–tocopherol (Toc; 300 μM) for 30 min and then treated with ginkgolide B (80 μM) for another 24 h. ROS generation was assayed using DCF-DA or DHR 123 dye (B). Apoptosis was evaluated using the Cell Death Detection ELISA kit (C). The data are representative of eight independent experiments. **P<0.01 and ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.


Figure 3.
Ginkgolide B triggers changes in intracellular calcium and NO content in MCF-7 cells. (A) Cells were incubated with ginkgolide B (GB) as indicated for 24 h. Intracellular Fluo-3 fluorescence intensity was measured in the presence/absence of extracellular Ca2+. (B) We examined the intracellular Ca2+ level changes triggered by treatment with ginkgolide B (GB; 80 μM), and the effects of ROS and NO inhibitors (NAC: 300 μM; L-NMMA: 400 μM; PTIO: 20 μM). (C) Cells were pretreated with L-NMMA (400 μM) or EGTA (400 μM) for 30 min and then incubated with ginkgolide B. Intracellular NO generation was measured by DAF-2DA fluorescence dye. Data are presented as percentage of control group. The data are representative of eight independent experiments. *P<0.05, **P<0.01 and ***P<0.001 versus the untreated control group; #P<0.001 versus the ginkgolide B-treated only group.
Figure 3.
Ginkgolide B triggers changes in intracellular calcium and NO content in MCF-7 cells. (A) Cells were incubated with ginkgolide B (GB) as indicated for 24 h. Intracellular Fluo-3 fluorescence intensity was measured in the presence/absence of extracellular Ca2+. (B) We examined the intracellular Ca2+ level changes triggered by treatment with ginkgolide B (GB; 80 μM), and the effects of ROS and NO inhibitors (NAC: 300 μM; L-NMMA: 400 μM; PTIO: 20 μM). (C) Cells were pretreated with L-NMMA (400 μM) or EGTA (400 μM) for 30 min and then incubated with ginkgolide B. Intracellular NO generation was measured by DAF-2DA fluorescence dye. Data are presented as percentage of control group. The data are representative of eight independent experiments. *P<0.05, **P<0.01 and ***P<0.001 versus the untreated control group; #P<0.001 versus the ginkgolide B-treated only group.

Figure 4.
Loss of MMP and activation of caspase-9 and -3 by ginkgolide B of MCF-7 cells. Cells were incubated with or without various concentrations of ginkgolide B (GB) for 24 h. (A) Mitochondrial membrane potential (MMP) changes were analyzed using 40 nM DiOC6(3) or 1 μM TMRE. (B) Caspase-9 activities were assayed using the Colorimetric Caspase-9 Assay kit. (C) Caspase-3 activities were analyzed using Z-DEVD-AFC as the substrate. Values are presented as means ± SD of six determinations. *P<0.05, **P<0.01 and ***P<0.001 versus the untreated control group.
Figure 4.
Loss of MMP and activation of caspase-9 and -3 by ginkgolide B of MCF-7 cells. Cells were incubated with or without various concentrations of ginkgolide B (GB) for 24 h. (A) Mitochondrial membrane potential (MMP) changes were analyzed using 40 nM DiOC6(3) or 1 μM TMRE. (B) Caspase-9 activities were assayed using the Colorimetric Caspase-9 Assay kit. (C) Caspase-3 activities were analyzed using Z-DEVD-AFC as the substrate. Values are presented as means ± SD of six determinations. *P<0.05, **P<0.01 and ***P<0.001 versus the untreated control group.


Figure 5.
NO involved in ginkgolide B-induced apoptotic biochemical changes in MCF-7 cells. Cells were pretreated with PTIO (20 μM) for 1 h and then treated with ginkgolide B (80 μM) for another 24 h. MMP change (A), activities of caspase-9 (B) and caspase-3 (C) were analyzed. Values are presented as means ± SD of eight determinations. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.
Figure 5.
NO involved in ginkgolide B-induced apoptotic biochemical changes in MCF-7 cells. Cells were pretreated with PTIO (20 μM) for 1 h and then treated with ginkgolide B (80 μM) for another 24 h. MMP change (A), activities of caspase-9 (B) and caspase-3 (C) were analyzed. Values are presented as means ± SD of eight determinations. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.


Figure 6.
Effect of NAC and PTIO on the mRNA expression levels of p53 and p21. Cells were pre-incubated with or without NAC (300 μM) and PTIO (20 μM) for 1 h and then treated with ginkgolide B (80 μM) for another 24 h. The mRNA expression levels of p53 (A) and p21 (B) were analyzed using real-time PCR. The given values are representative of eight determinations. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated group.
Figure 6.
Effect of NAC and PTIO on the mRNA expression levels of p53 and p21. Cells were pre-incubated with or without NAC (300 μM) and PTIO (20 μM) for 1 h and then treated with ginkgolide B (80 μM) for another 24 h. The mRNA expression levels of p53 (A) and p21 (B) were analyzed using real-time PCR. The given values are representative of eight determinations. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated group.

Figure 7.
Knockdown of p53 can protect MCF-7 cells against ginkgolide B-induced apoptosis. Cells were transfected with siRNA targeting p53, incubated for 24 h, and then treated with ginkgolide B (80 μM) for another 24 h. (A) The mRNA expression levels of p53 (A) and p21 (B) were analyzed using real-time PCR. (B) Cell apoptosis was measured as described in Figure 1. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.
Figure 7.
Knockdown of p53 can protect MCF-7 cells against ginkgolide B-induced apoptosis. Cells were transfected with siRNA targeting p53, incubated for 24 h, and then treated with ginkgolide B (80 μM) for another 24 h. (A) The mRNA expression levels of p53 (A) and p21 (B) were analyzed using real-time PCR. (B) Cell apoptosis was measured as described in Figure 1. ***P<0.001 versus the untreated control group. #P<0.001 versus the ginkgolide B-treated only group.

Figure 8.
Scheme of events occurring during ginkgolide B-induced cell apoptosis in MCF-7 cells.

Acknowledgements
This work was supported by grants (NSC95-2311-B-033-001-MY3 and NSC96-2627-M-033-002) from the National Science Council of Taiwan, ROC.
References and Notes
- Ahlemeyer, B.; Krieglstein, J. Pharmacological studies supporting the therapeutic use of Ginkgo biloba extract for Alzheimer’s disease. Pharmacopsychiatry 2003, 36 Suppl 1, S8–14. [Google Scholar]
- DeFeudis, F. V.; Papadopoulos, V.; Drieu, K. Ginkgo biloba extracts and cancer: a research area in its infancy. Fundam. Clin. Pharmacol 2003, 17, 405–417. [Google Scholar]
- Bate, C.; Salmona, M.; Williams, A. Ginkgolide B inhibits the neurotoxicity of prions or amyloid-beta1–42. J. Neuroinflammation 2004, 1, 4. [Google Scholar]
- Maitra, I.; Marcocci, L.; Droy-Lefaix, M. T.; Packer, L. Peroxyl radical scavenging activity of Ginkgo biloba extract EGb 761. Biochem. Pharmacol 1995, 49, 1649–1655. [Google Scholar]
- Pincemail, J.; Thirion, A.; Dupuis, M.; Braquet, P.; Drieu, K.; Deby, C. Ginkgo biloba extract inhibits oxygen species production generated by phorbol myristate acetate stimulated human leukocytes. Experientia 1987, 43, 181–184. [Google Scholar]
- Shen, J. G.; Zhou, D. Y. Efficiency of Ginkgo biloba extract (EGb 761) in antioxidant protection against myocardial ischemia and reperfusion injury. Biochem Mol. Biol. Int 1995, 35, 125–134. [Google Scholar]
- Pietri, S.; Maurelli, E.; Drieu, K.; Culcasi, M. Cardioprotective and anti-oxidant effects of the terpenoid constituents of Ginkgo biloba extract (EGb 761). J. Mol. Cell Cardiol 1997, 29, 733–742. [Google Scholar]
- Luo, Y.; Smith, J. V.; Paramasivam, V.; Burdick, A.; Curry, K. J.; Buford, J. P.; Khan, I.; Netzer, W. J.; Xu, H.; Butko, P. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12197–12202. [Google Scholar]
- Kim, K. S.; Rhee, K. H.; Yoon, J. H.; Lee, J. G.; Lee, J. H.; Yoo, J. B. Ginkgo biloba extract (EGb 761) induces apoptosis by the activation of caspase-3 in oral cavity cancer cells. Oral. Oncol 2005, 41, 383–389. [Google Scholar]
- Thompson, C. B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar]
- Halliwell, B.; Gutteridge, J. M. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol 1990, 186, 1–85. [Google Scholar]
- Chan, W. H. Ginkgolide B induces apoptosis and developmental injury in mouse embryonic stem cells and blastocysts. Hum. Reprod 2006, 21, 2985–2995. [Google Scholar]
- Chan, W. H.; Shiao, N. H.; Lu, P. Z. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol. Lett 2006, 167, 191–200. [Google Scholar]
- Martins, L. M.; Kottke, T.; Mesner, P. W.; Basi, G. S.; Sinha, S.; Frigon, N., Jr; Tatar, E.; Tung, J. S.; Bryant, K.; Takahashi, A.; Svingen, P. A.; Madden, B. J.; McCormick, D. J.; Earnshaw, W. C.; Kaufmann, S. H. Activation of multiple interleukin-1beta converting enzyme homologues in cytosol and nuclei of HL-60 cells during etoposide-induced apoptosis. J. Biol. Chem 1997, 272, 7421–7430. [Google Scholar]
- Nicholson, D. W.; Thornberry, N. A. Caspases: killer proteases. Trends Biochem. Sci 1997, 22, 299–306. [Google Scholar]
- Tsujimoto, Y.; Shimizu, S. Bcl-2 family: life-or-death switch. FEBS Lett 2000, 466, 6–10. [Google Scholar]
- Ekmekcioglu, S.; Tang, C. H.; Grimm, E. A. NO news is not necessarily good news in cancer. Curr. Cancer Drug Targets 2005, 5, 103–115. [Google Scholar]
- Zhou, J.; Brune, B. NO and transcriptional regulation: from signaling to death. Toxicology 2005, 208, 223–233. [Google Scholar]
- Rao, C. V. Nitric oxide signaling in colon cancer chemoprevention. Mutat. Res 2004, 555, 107–119. [Google Scholar]
- Lu, Z.; Tao, Y.; Zhou, Z.; Zhang, J.; Li, C.; Ou, L.; Zhao, B. Mitochondrial reactive oxygen species and nitric oxide-mediated cancer cell apoptosis in 2-butylamino-2-demethoxyhypocrellin B photodynamic treatment. Free Radic. Biol. Med 2006, 41, 1590–1605. [Google Scholar]
- Nazarewicz, R. R.; Zenebe, W. J.; Parihar, A.; Larson, S. K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res 2007, 67, 1282–1290. [Google Scholar]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci 2005, 26, 190–195. [Google Scholar]
- Brookes, P. S. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar]
- Dennis, J.; Bennett, J. P., Jr. Interactions among nitric oxide and Bcl-family proteins after MPP+ exposure of SH-SY5Y neural cells I: MPP+ increases mitochondrial NO and Bax protein. J. Neurosci. Res 2003, 72, 76–88. [Google Scholar]
- Elfering, S. L.; Sarkela, T. M.; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem 2002, 277, 38079–38086. [Google Scholar]
- Dedkova, E. N.; Ji, X.; Lipsius, S. L.; Blatter, L. A. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol 2004, 286, C406–415. [Google Scholar]
- Chan, W. H. Effect of resveratrol on high glucose-induced stress in human leukemia K562 cells. J. Cell. Biochem 2005, 94, 1267–1279. [Google Scholar]
- Hsuuw, Y. D.; Chang, C. K.; Chan, W. H.; Yu, J. S. Curcumin prevents methylglyoxal-induced oxidative stress and apoptosis in mouse embryonic stem cells and blastocysts. J. Cell. Physiol 2005, 205, 379–386. [Google Scholar]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 3100–3105. [Google Scholar]
- Webster, R. P.; Gawde, M. D.; Bhattacharya, R. K. Protective effect of rutin, a flavonol glycoside, on the carcinogen-induced DNA damage and repair enzymes in rats. Cancer Lett 1996, 109, 185–191. [Google Scholar]
- Mutoh, M.; Takahashi, M.; Fukuda, K.; Matsushima-Hibiya, Y.; Mutoh, H.; Sugimura, T.; Wakabayashi, K. Suppression of cyclooxygenase-2 promoter-dependent transcriptional activity in colon cancer cells by chemopreventive agents with a resorcin-type structure. Carcinogenesis 2000, 21, 959–963. [Google Scholar]
- Chan, W. H. Ginkgolides induce apoptosis and decrease cell numbers in mouse blastocysts. Biochem. Biophys. Res. Commun 2005, 338, 1263–1267. [Google Scholar]
- Buttke, T. M.; Sandstrom, P. A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar]
- Slater, A. F.; Nobel, C. S.; Maellaro, E.; Bustamante, J.; Kimland, M.; Orrenius, S. Nitrone spin traps and a nitroxide antioxidant inhibit a common pathway of thymocyte apoptosis. Biochem. J 1995, 306 Pt 3, 771–778. [Google Scholar]
- Chan, W. H.; Wu, C. C.; Yu, J. S. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. J. Cell. Biochem 2003, 90, 327–338. [Google Scholar]
- Long, L. H.; Clement, M. V.; Halliwell, B. Artifacts in cell culture: rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem. Biophys. Res. Commun 2000, 273, 50–53. [Google Scholar]
- Halliwell, B. Oxidative stress in cell culture: an under-appreciated problem? FEBS Lett 2003, 540, 3–6. [Google Scholar]
- Kroemer, G.; Zamzami, N.; Susin, S. A. Mitochondrial control of apoptosis. Immunol. Today 1997, 18, 44–51. [Google Scholar]
- Green, D. R.; Reed, J. C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Liu, G.; Hinch, B.; Davatol-Hag, H.; Lu, Y.; Powers, M.; Beavis, A. D. Temperature dependence of the mitochondrial inner membrane anion channel. The relationship between temperature and inhibition by protons. J. Biol. Chem 1996, 271, 19717–19723. [Google Scholar]
- Almeida, R. D.; Manadas, B. J.; Carvalho, A. P.; Duarte, C. B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar]
- Inanami, O.; Yoshito, A.; Takahashi, K.; Hiraoka, W.; Kuwabara, M. Effects of BAPTA-AM and forskolin on apoptosis and cytochrome c release in photosensitized Chinese hamster V79 cells. Photochem. Photobiol 1999, 70, 650–655. [Google Scholar]
- Monteiro, H. P.; Silva, E. F.; Stern, A. Nitric oxide: a potential inducer of adhesion-related apoptosis--anoikis. Nitric Oxide 2004, 10, 1–10. [Google Scholar]
- Li, C. Q.; Wogan, G. N. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005, 226, 1–15. [Google Scholar]
- Gomes, E. R.; Almeida, R. D.; Carvalho, A. P.; Duarte, C. B. Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism. Photochem. Photobiol 2002, 76, 423–430. [Google Scholar]
- Li, C. Q.; Robles, A. I.; Hanigan, C. L.; Hofseth, L. J.; Trudel, L. J.; Harris, C. C.; Wogan, G. N. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res 2004, 64, 3022–3029. [Google Scholar]
- Okada, H.; Mak, T. W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar]
- Chan, W. H.; Wu, H. J. Methylglyoxal and high glucose co-treatment induces apoptosis or necrosis in human umbilical vein endothelial cells. J. Cell. Biochem 2007. In Press. [Google Scholar]
- Chan, W. H.; Hsuuw, Y. D. Dosage effects of ginkgolide B on ethanol-induced cell death in human hepatoma G2 cells. Ann. N. Y. Acad. Sci 2007, 1095, 388–398. [Google Scholar]
- Chan, W. H. Citrinin induces apoptosis via a mitochondria-dependent pathway and inhibition of survival signals in embryonic stem cells, and causes developmental injury in blastocysts. Biochem. J 2007, 404, 317–326. [Google Scholar]
- Chan, W. H.; Chang, Y. J. Dosage effects of resveratrol on ethanol-induced cell death in the human K562 cell line. Toxicol. Lett 2006, 161, 1–9. [Google Scholar]
- Hsieh, Y. J.; Wu, C. C.; Chang, C. J.; Yu, J. S. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell. Physiol 2003, 194, 363–375. [Google Scholar]
- Aoshima, H.; Satoh, T.; Sakai, N.; Yamada, M.; Enokido, Y.; Ikeuchi, T.; Hatanaka, H. Generation of free radicals during lipid hydroperoxide-triggered apoptosis in PC12h cells. Biochim. Biophys. Acta 1997, 1345, 35–42. [Google Scholar]
- Nakatsubo, N.; Kojima, H.; Kikuchi, K.; Nagoshi, H.; Hirata, Y.; Maeda, D.; Imai, Y.; Irimura, T.; Nagano, T. Direct evidence of nitric oxide production from bovine aortic endothelial cells using new fluorescence indicators: diaminofluoresceins. FEBS Lett 1998, 427, 263–266. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Chan, W.-H. The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2007, 8, 1177-1195. https://doi.org/10.3390/i8111177
AMA Style
Chan W-H. The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2007; 8(11):1177-1195. https://doi.org/10.3390/i8111177
Chicago/Turabian StyleChan, Wen-Hsiung. 2007. "The Signaling Cascades of Ginkgolide B-Induced Apoptosis in MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 8, no. 11: 1177-1195. https://doi.org/10.3390/i8111177