Calorimetric and Computational Study of Enthalpy of Formation of Diperoxide of Cyclohexanone

Abstract
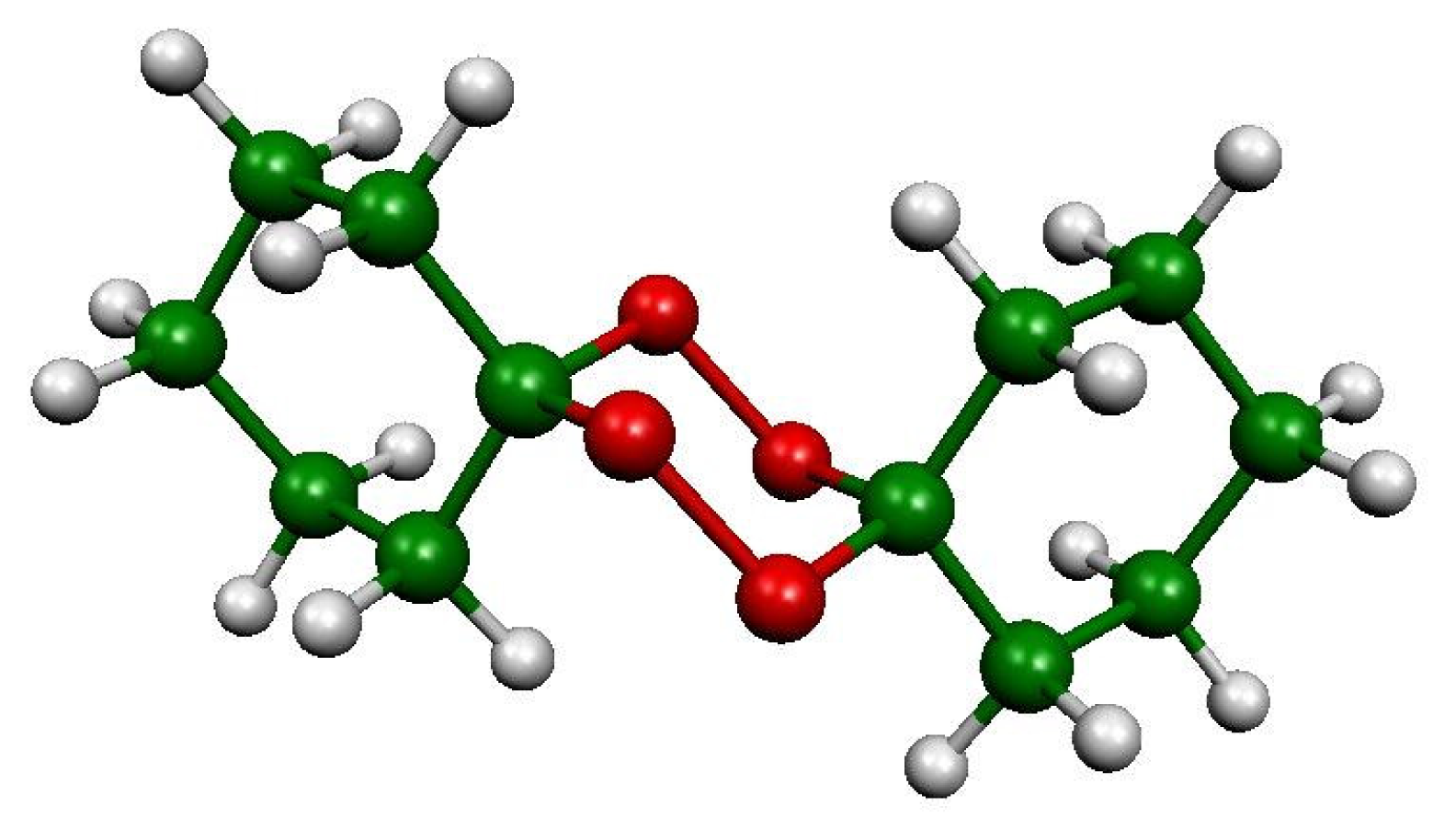
:1. Introduction
2. Results and Discussion
2.1. Experimental enthalpy of formation
2.2. Theoretical calculation of the enthalpy of formation
3. Experimental Section
3.1. Material
3.2. Thermochemical measurements
3.3. Theoretical calculations
4. Conclusions


{kind=link}
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|
| MDPCH/g | 0.0008 | 0.0005 | 0.0010 | 0.0026 | 0.0016 | 0.0006 | 0.0007 | 0.0011 |
| mgel/g | 0.0383 | 0.0405 | 0.0391 | 0.0363 | 0.0385 | 0.0385 | 0.0414 | 0.0414 |
| mFe/g | 0.0092 | 0.0093 | 0.0096 | 0.0083 | 0.0087 | 0.0075 | 0.0112 | 0.0176 |
| ΔT/K | 0.05620 | 0.056515 | 0.05604 | 0.05514 | 0.05599 | 0.05315 | 0.05923 | 0.06408 |
| (mwater+E)ΔT/Ja | 809.1952 | 840.8873 | 833.7457 | 820.4288 | 833.1059 | 790.7455 | 881.2692 | 953.4474 |
| mgel ΔUgel/Jb | 698.4167 | 738.5346 | 713.0051 | 661.9456 | 702.0635 | 702.0635 | 754.9463 | 754.9463 |
| mFe ΔUFe/Jc | 85.8389 | 86.7720 | 89.5711 | 77.4417 | 81.1738 | 69.9774 | 104.4996 | 164.2136 |
| ΔUc/J/gd | 31174.641 | 31162.118 | 31169.624 | 31169.808 | 31167.729 | 31173.660 | 31176.03 | 31170.256 |
| ΔHc/kJ/mol | −7115.27 | −7112.38 | −7114.10 | −7114.14 | −7113.68 | −7115.02 | −7115.56 | −7114.26 |
| Experience Nº | a joule/mol | b kJ/mol | c kJ/mol | d kJ/mol |
|---|---|---|---|---|
| 1 | 31174.641 | −7115.27 | −465.18 | −402.84 |
| 2 | 31162.118 | −7112.38 | −468.02 | −405.68 |
| 3 | 31169.624 | −7114.10 | −466.31 | −403.97 |
| 4 | 31169.808 | −7114.14 | −466.26 | −403.93 |
| 5 | 31167.729 | −7113.68 | −466.73 | −404.38 |
| 6 | 31173.660 | −7115.02 | −465.39 | −403.04 |
| 7 | 31176.030 | −7115.56 | −464.84 | −402.50 |
| 8 | 31170.256 | −7114.26 | −466.18 | −403.84 |
| Average value | 31170.483 | −7114.31 | −466.10 | −403.76 |
| Standard deviation | 1.057055 | 0.241008 | 0.241008 | 0.241008 |
| - (ɛ0 + Hcorr)a [kJ/mol] | ||||||
|---|---|---|---|---|---|---|
| AM1 | PM3 | RHF 3–21G | RHF 6–31G | B3LYP 3–21G | B3LYP 6–31G | |
| DPCH | −560.73 | −524.10 | 1997480.21 | 2007620.97 | 2010027.54 | 2020385.03 |
| CH4 | −89.49 | −74.38 | 104823.31 | 105358.23 | 105682.13 | 106231.30 |
| CH | −307.08 | −325.32 | 611026.75 | 614161.77 | 615492.77 | 618683.31 |
| CH3OOCH3 | −128.30 | −85.63 | 597260.18 | 600286.20 | 600769.65 | 603880.54 |
| ΔHrb | −47.91 | 0.28355 | 201.83 | 157.97 | 231.20 | 182.54 |
| a [kJ mol−1]
| |||||
|---|---|---|---|---|---|
| AM1 | PM3 | RHF 3–21G | RHF 6–31G | B3LYP 3–21G | B3LYP 6–31G |
| −170.280 | −200.20 | −402.30 | −358.17 | −431.40 | −382.74 |
Acknowledgements
References and Notes
- a)McCullough, K.J.; Morgan, A.R.; Nonhebel, D.C.; Pauson, P.L.; White, G.J. Ketones-derived Peroxides. Part. III. Decompositions of Cyclic Peroxides derived from Dialkyl Ketones. J. Chem. Res. Synop 1980, 2. [Google Scholar]McCullough, K.J.; Morgan, A.R.; Nonhebel, D.C.; Pauson, P.L. Ketones-derived Peroxides. Part. I. Synthetic Methods. J. Chem. Res. Synop 1981, 2, 34–36. [Google Scholar]
- Leiva, L.C.A.; Romero, J.M.; Jorge, N.L.; Gómez Vara, M.E.; Cafferata, L.F.R. Síntesis y Descomposición Térmica Del Diperóxido De Formaldehído. Revista Internacional Información Tecnológica 2002, 13(2), 23–26. [Google Scholar]
- Adam, W.; Hadjiarapoglou, L.P.; Curci, R.; Mello, R. Organic Peroxides; Ando, W., Ed.; John Wiley & Sons: Chichester, England, 1992; Volume Chapter 4. [Google Scholar]
- Story, P.R.; Busch, P. Advances in Organic Chemistry; Taylor, E.C., Ed.; Wiley: New York, 1972; Volume 8, pp. 67–95. [Google Scholar]
- Eyler, G.N.; Mateo, C.M.; Alvarez, E.E.; Cañizo, A.I. Termal Decomposition reaction of Acetone Triperoxide in Toluene solution. J. Org. Chem. 2000, 65, 2319–2321. [Google Scholar]
- Eyler, G.N.; Mateo, C.M.; Álvarez, E.E.; Cañizo, A.I.; Cafferata, L.F.R. Solvent Effect in the Thermal Decomposition Reaction of trans-3,3-Dimethyl-5,6-tetramethylene-1,2,4-trioxacyclohexane. J. Org. Chem. 1999, 64, 8457–8460. [Google Scholar]
- Morales, G.; Eyler, G.N.; Cerna, J.R.; Cañizo, A.I. Use of Cyclic Di- and Triperoxides as Initiators of Styrene Polymerization at High Temperature with a View to Their Use in Industrial Applications. Molecules 2000, 5, 549–550. [Google Scholar]
- Vennerstrom, J.L.; Fu, H.N.; Ellis, W.Y.; Ager, A.L., Jr.; Wood, J.K.; Andersen, S.L.; Gerena, L.; Milhous, W.K. Dispiro-1,2,4,5-tetraoxanes: a new class of antimalarial peroxides. J. Medical Chem 1992, 35, 3023–3027. [Google Scholar]
- Kim, H.S.; Shibata, Y.; Wataya, Y.; Tsuchiya, K.; Masuyama, A.; Nojima, M. Synthesis and Antimalarial Activity of Cyclic Peroxides, 1,2,4,5,7-Pentoxocanes and 1,2,4,5-Tetroxanes. J. Med. Chem 1999, 42, 2604–2609. [Google Scholar]
- McCullogh, K.; Nonami, Y.; Masuyama, A.; Nojima, M.; Kim, HS; Wataya, Y. Synthesis, crystal structure and antimalarial activity of novel 1,2,5,6-tetraoxacycloalkanes from 2,3-dihydroperoxy-2-phenylnorbornane. Tetrahedrom Lett. 1999, 40, 9151–9155. [Google Scholar]
- Rogers, D.W. Computational Chemistry Using the PC, Third Edition ed; Wiley: New York, 2003. [Google Scholar]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A New General Purpose Quantum Mechanical Model. J. Am. Chem. Soc 1985, 107, 3902–3909. [Google Scholar]
- Stewart, J.J.P. Semiempirical Molecular Orbital Methods. In Reviews in Computational Chemistry; Volume 1, Lipkpwitz, K.B., Boyd, D.B., Eds.; VCH: New York, 1990; pp. 45–81. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem 1989, 10, 209–220. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem 1989, 10, 221–264. [Google Scholar]
- Höltje, H.D.; Sippl, W.; Rognan, D.; Folkers, G. Molecular Modeling. In Basic Principles and Applications, Second Edition ed; Wiley: New York, 2003; p. 23. [Google Scholar]
- Story, P.R.; Lee, B.; Bishop, C.E.; Denson, D.D.; Busch, P. Macrocyclic Synthesis. II. Cyclohexanone Peroxides. J. Org. Chem 1970, 35, 3059–3062. [Google Scholar]
- Hubbart, W.N.; Scott, D.W.; Waddington, G. Experimental Thermochemistry; Rossini, F.D., Ed.; Interscience: New York, 1956; pp. 75–127. [Google Scholar]
- Cox, J.D.; Wagman, D.D.; Medvedev, V.A. (Eds.) CODATA Key Values for Thermodynamics; Hemisphere: New York, 1989.
- Wieser, M.E. Atomic Weights of The Elements 2005 (IUPAC Technical Report). Pure Appl. Chem. 2006, 78(11), 2051–2066. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.M.W.; Johnson, B.G.; Robb, A.; Cheeseman, J.R.; Keith, T.A.; Petersson, G.A.; Montgomery, J.A.; Kaghavachari, K.; Al-Laham, M.A.; Kakrewski, V.G.; Ortiz, J.V.; Foresman, J.B.; Cioslowski, J.; Stefanov, B.B.; Nanayakkara, A.; Challacombe, M.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andrés, J.L.; Replogle, E.S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J.S.; DeFrees, D.J.; Baker, J.; Stewart, J.P.; Head-Gordon, M.; González, C.; Pople, J. A. Gaussian 94; rev D3, SGI; Gaussian, Inc: Pittsburgh, PA, 1995. [Google Scholar]
© 2007 by MDPI ( http://www.mdpi.org) Reproduction is permitted for noncommercial purposes.
Share and Cite
Romero, J.M.; Bustillo, S.; Maisuls, H.E.R.; Jorge, N.L.; Vara, M.E.G.; Castro, E.A.; Jubert, A.H. Calorimetric and Computational Study of Enthalpy of Formation of Diperoxide of Cyclohexanone. Int. J. Mol. Sci. 2007, 8, 688-694. https://doi.org/10.3390/i8070688
Romero JM, Bustillo S, Maisuls HER, Jorge NL, Vara MEG, Castro EA, Jubert AH. Calorimetric and Computational Study of Enthalpy of Formation of Diperoxide of Cyclohexanone. International Journal of Molecular Sciences. 2007; 8(7):688-694. https://doi.org/10.3390/i8070688
Chicago/Turabian StyleRomero, Jorge Marcelo, Soledad Bustillo, Hugo Enrique Ramirez Maisuls, Nelly Lidia Jorge, Manuel Eduardo Gómez Vara, Eduardo Alberto Castro, and Alicia H. Jubert. 2007. "Calorimetric and Computational Study of Enthalpy of Formation of Diperoxide of Cyclohexanone" International Journal of Molecular Sciences 8, no. 7: 688-694. https://doi.org/10.3390/i8070688