Substituting Nε-thioacetyl-lysine for Nε-acetyl-lysine in Peptide Substrates as a General Approach to Inhibiting Human NAD+-dependent Protein Deacetylases

Abstract
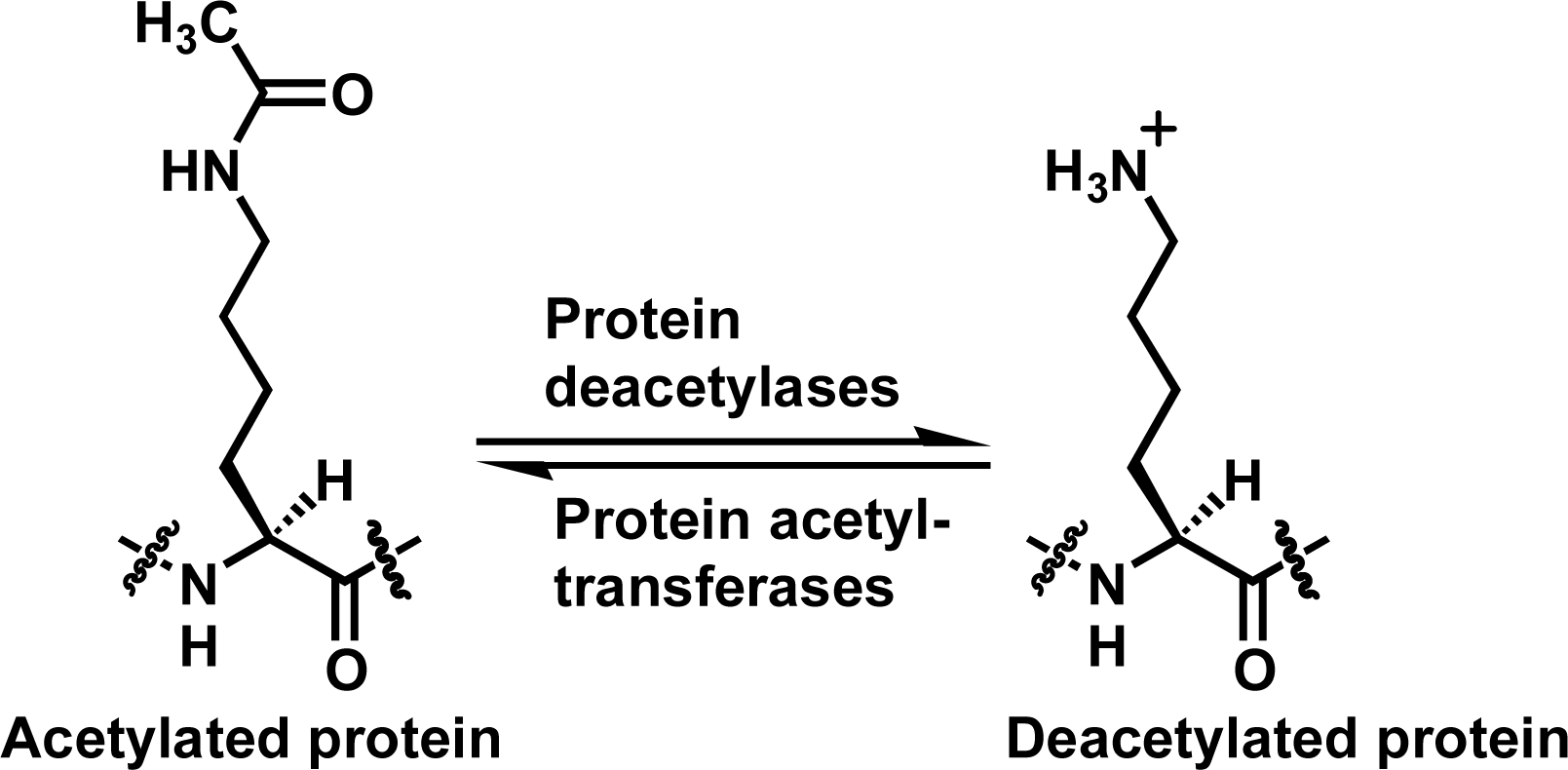
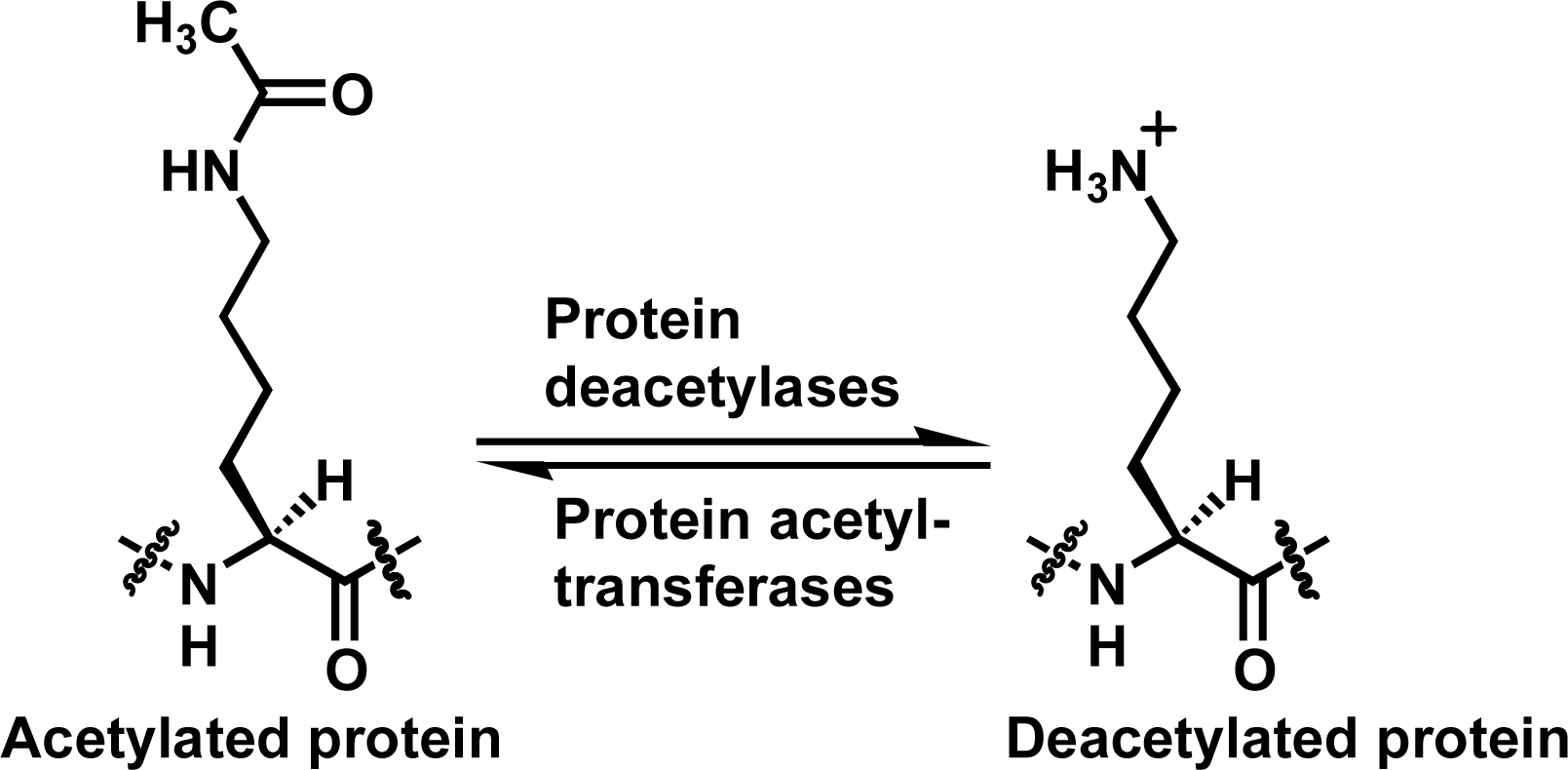
:1. Introduction
2. Results and Discussion
2.1 Peptide-based potent and selective inhibitors of SIRT1, SIRT2, and SIRT3
2.2 Possible HDAC8-catalyzed dethioacetylation of peptide-based inhibitors
3. Experimental Section
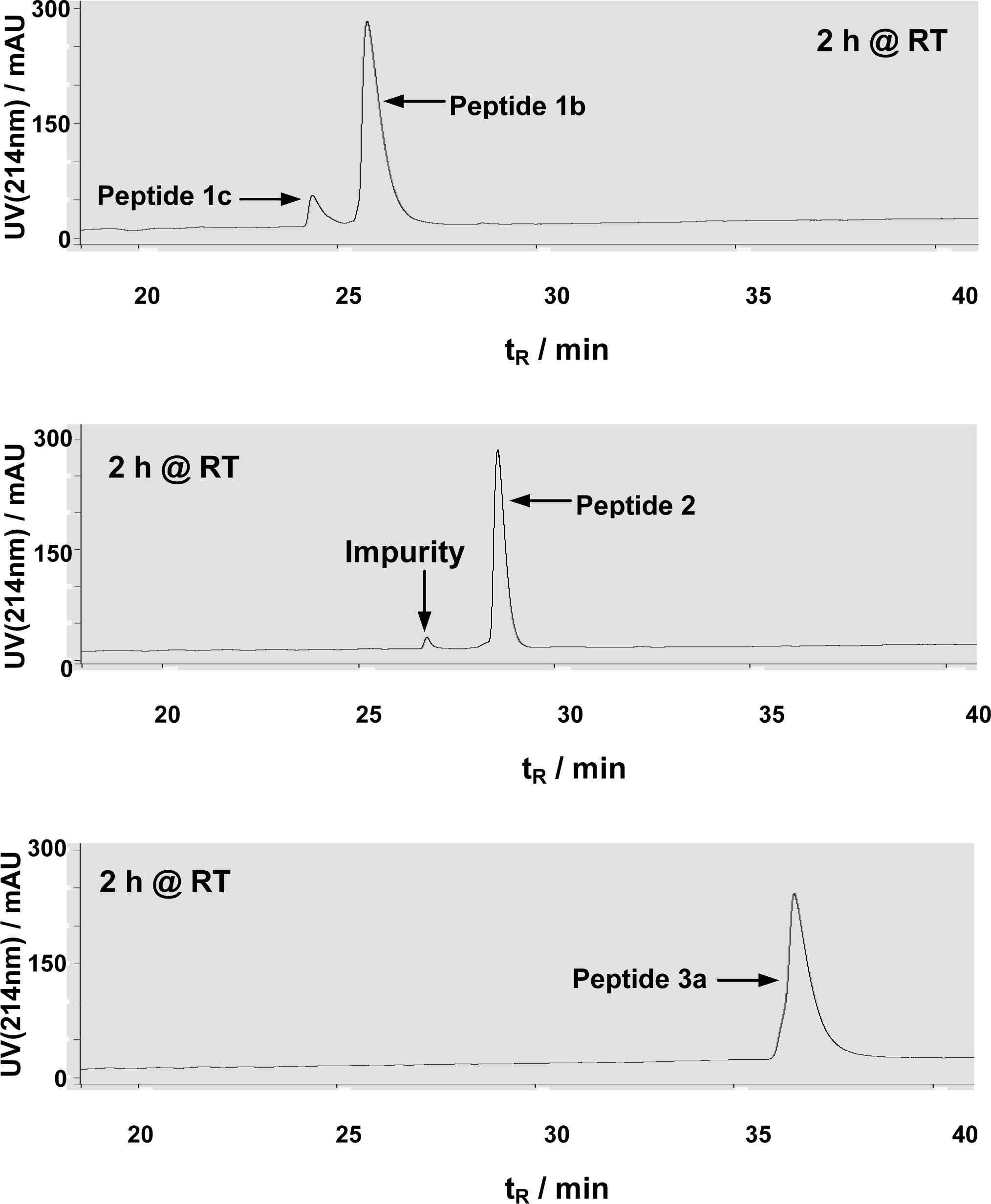
3.1 Peptide synthesis and purification
3.2 Inhibition assays with recombinant SIRT1, SIRT2, and SIRT3
3.3 Assay with recombinant HDAC8
4. Conclusions
Acknowledgements
References
- Yang, XJ; Seto, E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar]
- Glozak, MA; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar]
- Hodawadekar, SC; Marmorstein, R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 2007, 26, 5528–5540. [Google Scholar]
- Batta, K; Das, C; Gadad, S; Shandilya, J; Kundu, TK. Reversible acetylation of non histone proteins: role in cellular function and disease. Subcell Biochem 2007, 41, 193–212. [Google Scholar]
- Shahbazian, MD; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007, 76, 75–100. [Google Scholar]
- Saunders, LR; Verdin, E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene 2007, 26, 5489–5504. [Google Scholar]
- Swaminathan, V; Reddy, BA; Ruthrotha Selvi, B; Sukanya, MS; Kundu, TK. Small molecule modulators in epigenetics: implications in gene expression and therapeutics. Subcell Biochem 2007, 41, 397–428. [Google Scholar]
- Marchion, D; Munster, P. Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther 2007, 7, 583–598. [Google Scholar]
- Xu, WS; Parmigiani, RB; Marks, PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar]
- Duvic, M; Vu, J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous Tcell lymphoma. Expert Opin Investig Drugs 2007, 16, 1111–1120. [Google Scholar]
- Guarente, L. Sirtuins as potential targets for metabolic syndrome. Nature 2006, 444, 868–874. [Google Scholar]
- Garske, AL; Smith, BC; Denu, JM. Linking SIRT2 to Parkinson's disease. ACS Chem Biol 2007, 2, 529–532. [Google Scholar]
- Gan, L. Therapeutic potential of sirtuin-activating compounds in Alzheimer's disease. Drug News Perspect 2007, 20, 233–239. [Google Scholar]
- Thiagalingam, S; Cheng, KH; Lee, HJ; Mineva, N; Thiagalingam, A; Ponte, JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci 2003, 983, 84–100. [Google Scholar]
- Gregoretti, IV; Lee, YM; Goodson, HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004, 338, 17–31. [Google Scholar]
- Napper, AD; Hixon, J; McDonagh, T; Keavey, K; Pons, JF; Barker, J; Yau, WT; Amouzegh, P; Flegg, A; Hamelin, E; Thomas, RJ; Kates, M; Jones, S; Navia, MA; Saunders, JO; DiStefano, PS; Curtis, R. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem 2005, 48, 8045–8054. [Google Scholar]
- Outeiro, TF; Kontopoulos, E; Altmann, SM; Kufareva, I; Strathearn, KE; Amore, AM; Volk, CB; Maxwell, MM; Rochet, JC; McLean, PJ; Young, AB; Abagyan, R; Feany, MB; Hyman, BT; Kazantsev, AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science 2007, 317, 516–519. [Google Scholar]
- Fatkins, DG; Monnot, AD; Zheng, W. Nε-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nε-deacetylation. Bioorg Med Chem Lett 2006, 16, 3651–3656. [Google Scholar]
- Denu, JM. The Sir 2 family of protein deacetylases. Curr Opin Chem Biol 2005, 9, 431–440. [Google Scholar]
- Jamonnak, N; Fatkins, DG; Wei, L; Zheng, W. N(epsilon)-Methanesulfonyl-lysine as a nonhydrolyzable functional surrogate for N(epsilon)-acetyl-lysine. Org Biomol Chem 2007, 5, 892–896. [Google Scholar]
- Sauve, AA; Celic, I; Avalos, J; Deng, H; Boeke, JD; Schramm, VL. Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry 2001, 40, 15456–15463. [Google Scholar]
- Smith, JS; Avalos, J; Celic, I; Muhammad, S; Wolberger, C; Boeke, JD. SIR2 family of NAD(+)-dependent protein deacetylases. Methods Enzymol 2002, 353, 282–300. [Google Scholar]
- Avalos, JL; Celic, I; Muhammad, S; Cosgrove, MS; Boeke, JD; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 2002, 10, 523–535. [Google Scholar]
- Wellings, DA; Atherton, E. Standard Fmoc protocols. Methods Enzymol 1997, 289, 44–67. [Google Scholar]
- Fatkins, DG; Zheng, W. A spectrophotometric assay for histone deacetylase 8. Anal Biochem 2008, 372, 82–88. [Google Scholar]
- Wadia, JS; Dowdy, SF. Transmembrane delivery of protein and peptide drugs by TATmediated transduction in the treatment of cancer. Adv Drug Deliv Rev 2005, 57, 579–596. [Google Scholar]
- Fuchs, SM; Raines, RT. Polyarginine as a multifunctional fusion tag. Protein Sci 2005, 14, 1538–1544. [Google Scholar]
- Rothbard, JB; Jessop, TC; Wender, PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev 2005, 57, 495–504. [Google Scholar]
- Wright, LR; Rothbard, JB; Wender, PA. Guanidinium rich peptide transporters and drug delivery. Curr Protein Pept Sci 2003, 4, 105–124. [Google Scholar]
- Zheng, Y; Balasubramanyam, K; Cebrat, M; Buck, D; Guidez, F; Zelent, A; Alani, RM; Cole, PA. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. J Am Chem Soc 2005, 127, 17182–17183. [Google Scholar]
- Avalos, JL; Celic, I; Muhammad, S; Cosgrove, MS; Boeke, JD; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol Cell 2002, 10, 523–535. [Google Scholar]
- Garske, AL; Denu, JM. SIRT1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry 2006, 45, 94–101. [Google Scholar]
- Langley, E; Pearson, M; Faretta, M; Bauer, UM; Frye, RA; Minucci, S; Pelicci, PG; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 2002, 21, 2383–2396. [Google Scholar]
- Bannister, AJ; Cook, A; Kouzarides, T. In vitro DNA binding activity of Fos/Jun and BZLF1 but not C/EBP is affected by redox changes. Oncogene 1991, 6, 1243–1250. [Google Scholar]
- North, BJ; Schwer, B; Ahuja, N; Marshall, B; Verdin, E. Preparation of enzymatically active recombinant class III protein deacetylases. Methods 2005, 36, 338–345. [Google Scholar]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem J 1953, 55, 170–171. [Google Scholar]
- Smith, BC; Denu, JM. Mechanism-based inhibition of sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 2007, 46, 14478–14486, Published on Web 11/21/2007. [Google Scholar]
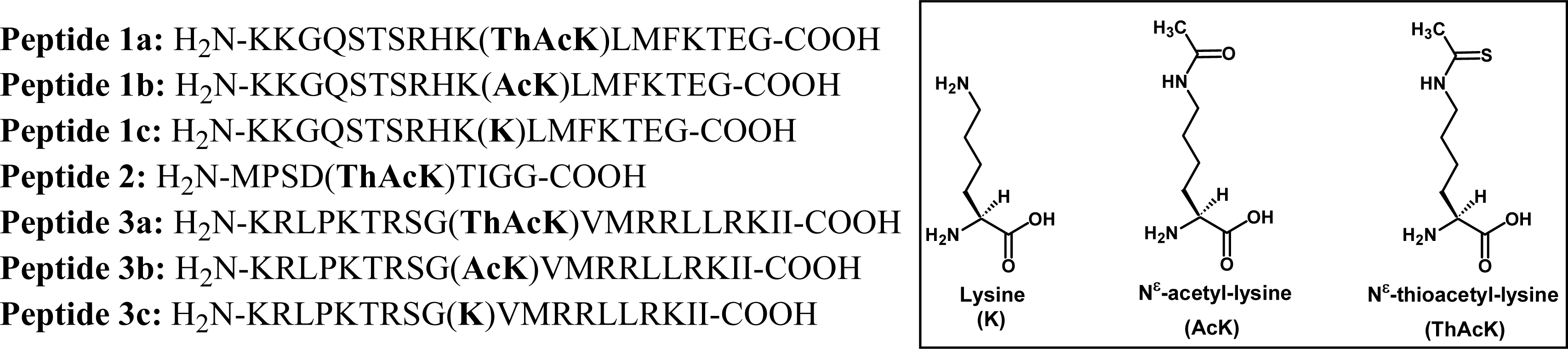


{kind=link}
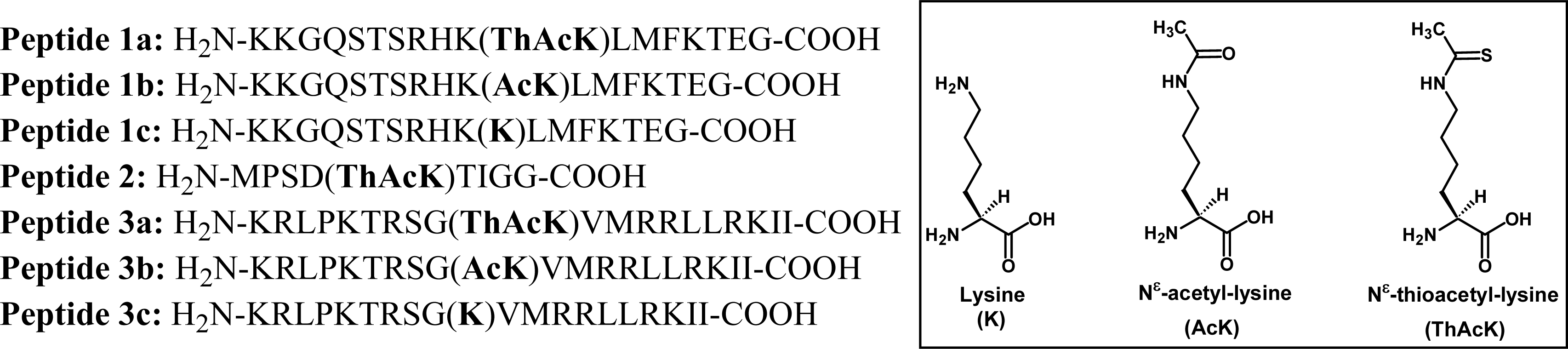{kind=link}
{kind=link}
| Peptide | IC50 (μM)b | HDAC8 | ||
|---|---|---|---|---|
| SIRT1 | SIRT2 | SIRT3 | ||
| 1a | 1.7 ± 0.4c | 1.8 ± 0.3 | 67.3 ± 2.4 | +c,d |
| 2 | 116.8 ± 12.0 | 11.4 ± 1.1 | 449.4 ± 18.4 | –e |
| 3a | 0.9 ± 0.2 | 4.3 ± 0.3 | 4.5 ± 2.0 | – |
Share and Cite
Fatkins, D.G.; Zheng, W. Substituting Nε-thioacetyl-lysine for Nε-acetyl-lysine in Peptide Substrates as a General Approach to Inhibiting Human NAD+-dependent Protein Deacetylases. Int. J. Mol. Sci. 2008, 9, 1-11. https://doi.org/10.3390/ijms9010001
Fatkins DG, Zheng W. Substituting Nε-thioacetyl-lysine for Nε-acetyl-lysine in Peptide Substrates as a General Approach to Inhibiting Human NAD+-dependent Protein Deacetylases. International Journal of Molecular Sciences. 2008; 9(1):1-11. https://doi.org/10.3390/ijms9010001
Chicago/Turabian StyleFatkins, David G., and Weiping Zheng. 2008. "Substituting Nε-thioacetyl-lysine for Nε-acetyl-lysine in Peptide Substrates as a General Approach to Inhibiting Human NAD+-dependent Protein Deacetylases" International Journal of Molecular Sciences 9, no. 1: 1-11. https://doi.org/10.3390/ijms9010001