Actomyosin Interaction: Mechanical and Energetic Properties in Different Nucleotide Binding States

Abstract
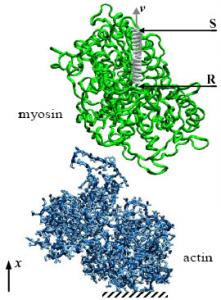
:
1. Introduction
2. Results and Discussion
2.1. Analysis of the Equilibrated Structures
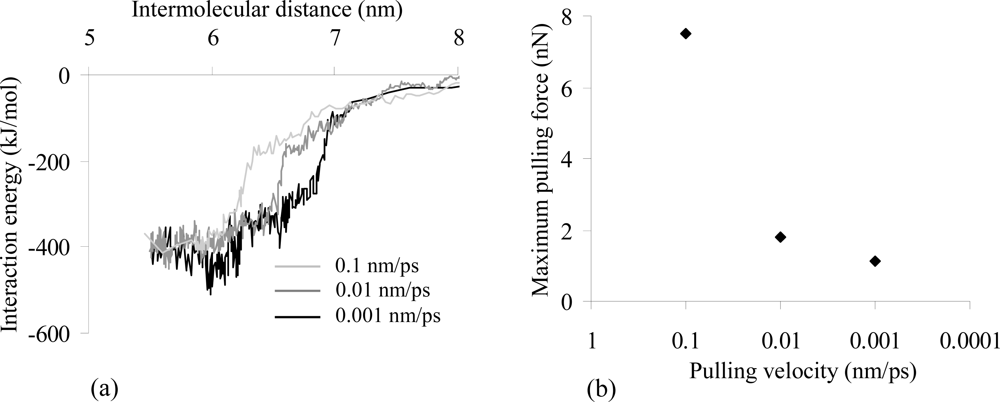
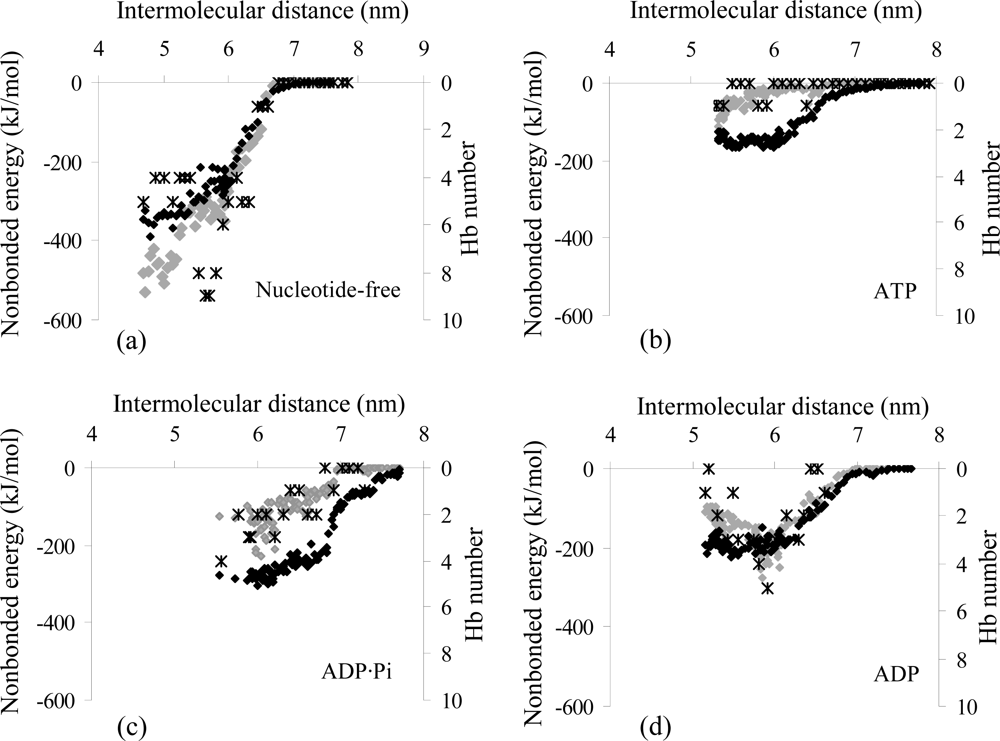
2.2. Analysis of the Interaction Properties of the Actomyosin Complex
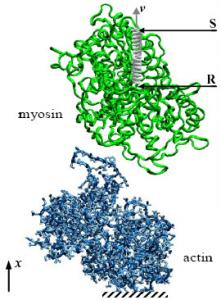
2.3. Influence of the Pulling Rate
2.4. Influence of the Spring Constant
2.5. Evaluation of the Interaction Forces Characterising the Actomyosin Complexes
3. Experimental Section
3.1. Molecular Models
3.2. Numerical Set-up
4. Conclusions
Acknowledgments
References
- Rayment, I; Rypniewski, WR; Schmidt-Bae, K; Smith, R; Tomchick, DR; Benning, MM; Winkelmann, DA; Wesenberg, G; Holden, HM. Three-Dimensional Structure of Myosin Subfragment-1: a Molecular Motor. Science 1993, 261, 50–58. [Google Scholar]
- Fisher, AJ; Smith, CA; Thoden, JB; Smith, R; Sutoh, K; Holden, HM; Rayment, I. X-Ray Structures of the Myosin Motor Domain of Dictyostelium Discoideum Complexed with MgADP.BeFx and MgADP.AlF4−. Biochemistry 1995, 34, 8960–8972. [Google Scholar]
- Smith, CA; Rayment, I. X-ray Structure of the Magnesium(II)-Pyrophosphate Complex of the Truncated Head of Dictyostelium Discoideum Myosin to 2.7 Å Resolution. Biochemistry 1995, 34, 8973–8981. [Google Scholar]
- Smith, CA; Rayment, I. X-ray Structure of the Magnesium(II).ADP.Vanadate Complex of the Dictyostelium discoideum Myosin Motor Domain to 1.9 Å Resolution. Biochemistry 1996, 35, 5404–5417. [Google Scholar]
- Bauer, CB; Kuhlman, PA; Bagshaw, CR; Rayment, I. X-ray Crystal Structure and Solution Fluorescence Characterization of Mg. 2′(3′)-O-(N-methylanthraniloyl) Nucleotides Bound to the Dictyostelium discoideum Myosin Motor Domain. J. Mol. Biol 1997, 274, 394–407. [Google Scholar]
- Gulick, AM; Bauer, CB; Thoden, JB; Rayment, I. Biochemistry 1997, 36, 11619–11628.
- Gulick, AM; Bauer, CB; Thoden, JB; Pate, E; Yount, RG; Rayment, I. X-ray Structures of the Dictyostelium discoideum Myosin Motor Domain with Six Non-nucleotide Analogs. J. Biol. Chem. 2000, 275, 398–408. [Google Scholar]
- Dominguez, R; Freyzon, Y; Trybus, KM; Cohen, C. Crystal Structure of a Vertebrate Smooth Muscle Myosin Motor Domain and its Complex with the Essential Light Chain: Visualization of the Pre-power Stroke State. Cell 1998, 94, 559–571. [Google Scholar]
- Houdusse, A; Kalabokis, VN; Himmel, D; Szent-Gyorgyi, AG; Cohen, C. Atomic Structure of Scallop Myosin Subfragment S1 Complexed with MgADP: A Novel Conformation of the Myosin Head. Cell 1999, 97, 459–470. [Google Scholar]
- Houdusse, A; Szent-Gyorgyi, AG; Cohen, C. Three Conformational States of Scallop Myosin S1. Proc. Natl. Acad. Sci. USA. 2000, 97, 11238–11243. [Google Scholar]
- Himmel, DM; Gourinath, S; Reshetnikova, L; Shen, Y; Szent-Gyorgyi, AG; Cohen, C. Crystallographic Findings on the Internally Uncoupled and Near-Rigor States of Myosin: Further Insights into the Mechanics of the Motor. Proc. Natl. Acad. Sci. USA. 2002, 99, 12645–12650. [Google Scholar]
- Gourinath, S; Himmel, DM; Brown, JH; Reshetnikova, L; Szent-Gyorgyi, AG; Cohen, C. Crystal Structure of Scallop Myosin S1 in the Pre-Power Stroke State to 2.6 Å Resolution: Flexibility and Function in the Head. Structure 2003, 11, 1621–1627. [Google Scholar]
- Risal, D; Gourinath, S; Himmel, DM; Szent-Gyorgyi, AG; Cohen, C. Myosin Subfragment 1 Structures Reveal a Partially Bound Nucleotide and a Complex Salt Bridge that Helps Couple Nucleotide and Actin Binding. Proc. Natl. Acad. Sci. USA. 2004, 101, 8930–8935. [Google Scholar]
- Bubb, MR; Govindasamy, L; Yarmola, EG; Vorobiev, SM; Almo, SC; Somasundaram, T; Chapman, MS; Agbandje-Mckenna, M; Mckenna, R. Polylysine Induces an Antiparallel Actin Dimer that Nucleates Filament Assembly: Crystal Structure at 3.5-A Resolution. J. Biol. Chem 2002, 277, 20999–21006. [Google Scholar]
- Otterbein, LR; Graceffa, P; Dominguez, R. The Crystal Structure of Uncomplexed Actin in the ADP State. Science 2001, 293, 708–711. [Google Scholar]
- Mclaughlin, PJ; Gooch, JT; Mannherz, HG; Weeds, AG. Structure of Gelsolin Segment 1-Actin Complex and the Mechanism of Filament Severing. Nature 1993, 364, 685–692. [Google Scholar]
- Choe, H; Burtnick, LD; Mejillano, M; Yin, HL; Robinson, RC; Choe, S. The Calcium Activation of Gelsolin: Insights from the 3A Structure of the G4-G6/Actin Complex. J. Mol. Biol. 2002, 324, 691–702. [Google Scholar]
- Kabsch, W; Mannherz, HG; Suck, D; Pai, EF; Holmes, KC. Atomic Structure of the Actin:Dnase I Complex. Nature 1990, 347, 37–44. [Google Scholar]
- Chen, LF; Winkler, H; Reedy, MK; Reedy, MC; Taylor, KA. Molecular Modeling of Averaged Rigor Crossbridges from Tomograms of Insect Flight Muscle. J. Struct. Biol 2002, 138, 92–104. [Google Scholar]
- Rayment, I; Holden, HM; Whittaker, M; Yohn, CB; Lorenz, M; Holmes, KC; Milligan, RA. Structure of the Actin-Myosin Complex and Its Implications for Muscle Contraction. Science 1993, 261, 58–65. [Google Scholar]
- Bauer, CB; Holden, HM; Thoden, JB; Smith, R; Rayment, I. X-Ray Structures of the Apo and MgATP-Bound States of Dictyostelium Discoideum Myosin Motor Domain. J. Biol. Chem. 2000, 275, 38494–38499. [Google Scholar]
- Li, G; Cui, Q. Mechanochemical Coupling in Myosin: A Theoretical Analysis with Molecular Dynamics and Combined QM/MM Reaction Path Calculations. J. Phys. Chem. B 2004, 108, 3342–3357. [Google Scholar]
- Yu, H; Ma, L; Yang, Y; Cui, Q. Mechanochemical Coupling in the Myosin Motor Domain. II. Analysis of Critical Residues. PLOS. Comput. Biol 2007, 3, 1–17. [Google Scholar]
- Kawakubo, T; Okada, O; Minami, T. Molecular Dynamics Simulations of Evolved Collective Motions of Atoms in the Myosin Motor Domain upon Perturbation of the ATPase Pocket. Biophys. Chem. 2005, 115, 77–85. [Google Scholar]
- Lawson, JD; Pate, E; Rayment, I; Yount, RG. Molecular Dynamics Analysis of Structural Factors Influencing Back Door Pi Release in Myosin. Biophys. J. 2004, 86, 3794–3803. [Google Scholar]
- Liu, Y; Scolari, M; Im, W; Woo, HJ. Protein-Protein Interactions in Actin-Myosin Binding and Structural Effects of R405Q Mutation: A Molecular Dynamics Study. Proteins 2006, 64, 156–166. [Google Scholar]
- Izrailev, S; Stepaniants, S; Isralewitz, B; Kosztin, D; Lu, H; Molnar, F; Wriggers, W; Schulten, K. Steered Molecular Dynamics. Computational Science and Engineering 1998, 39–65. [Google Scholar]
- Isralewitz, B; Gao, M; Schulten, K. Steered Molecular Dynamics and Mechanical Functions of Proteins. Curr. Opin. Struct. Biol 2001, 11, 224–230. [Google Scholar]
- Laskowski, RA; Macarthur, MW; Moss, DS; Thornton, JM. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. App. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- Nishizaka, T; Miyata, H; Yoshikawa, H; Ishiwata, S; Kinosita, K. Unbinding Force of a Single Motor Molecule of Muscle Measured Using Optical Tweezers. Nature 1995, 377, 251–253. [Google Scholar]
- Nishizaka, T; Miyata, H; Yoshikawa, H; Ishiwata, S; Kinosita, K. Mechanical Properties of Single Protein Motor of Muscle Studied by Optical Tweezers. Biophys. J. 1995, 68, 75b. [Google Scholar]
- Molloy, JE; Burns, JE; Sparrow, JC; Tregear, RT; Kendrick-Jones, J; White, DC. Single-Molecule Mechanics of Heavy Meromyosin and S1 Interacting with Rabbit or Drosophila Actins Using Optical Tweezers. Biophys. J. 1995, 68, 298S–305S. [Google Scholar]
- Nakajima, H; Kunioka, Y; Nakano, K; Shimizu, K; Seto, M; Ando, T. Scanning Force Microscopy of the Interaction Events between a Single Molecule of Heavy Meromyosin and Actin. Biochem. Bioph. Res. Co 1997, 234, 178–182. [Google Scholar]
- Merkel, R; Nassoy, P; Leung, A; Ritchie, K; Evans, E. Energy Landscapes of Receptor-Ligand Bonds Explored with Dynamic Force Spectroscopy. Nature 1999, 397, 50–53. [Google Scholar]
- Fritz, J; Katopodis, AG; Kolbinger, F; Anselmetti, D. Force-Mediated Kinetics of Single P-Selectin-Ligand Complexes Observed by Atomic Force Microscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 12283–12288. [Google Scholar]
- Soncini, M; Vesentini, S; Ruffoni, D; Orsi, M; Deriu, MA; Redaelli, A. Mechanical Response and Conformational Changes of Alpha-Actinin Domains during Unfolding: A Molecular Dynamics Study. Biomechan. Model Mechanobiol 2007, 6, 399–407. [Google Scholar]
- Deriu, MA; Enemark, S; Soncini, M; Montevecchi, FM; Redaelli, A. Tubulin: from Atomistic Structure to Supramolecular Mechanical Properties. J. Mater. Sci. 2007, 42, 8864–8872. [Google Scholar]
- Lenne, PF; Raae, AJ; Altmann, SM; Saraste, M; Horber, JKH. States and Transitions during Forced Unfolding of a Single Spectrin Repeat. FEBS. Lett. 2000, 476, 124–128. [Google Scholar]
- Lorenzo, AC; Bisch, PM. Analyzing Different Parameters of Steered Molecular Dynamics for Small Membrane Interacting Molecules. J. Mol. Graph. Model 2005, 24, 59–71. [Google Scholar]
- Holmes, KC; Angert, I; Kull, FJ; Jahn, W; Schroder, RR. Electron Cryo-microscopy Shows how Strong Binding of Myosin to Actin Releases Nucleotide. Nature 2003, 425, 423–427. [Google Scholar]
- Finer, JT; Simmons, RM; Spudich, JA. Single Myosin Molecule Mechanics: Piconewton Forces and Nanometre Steps. Nature 1994, 368, 113–119. [Google Scholar]
- Izrailev, S; Stepaniants, S; Balsera, M; Oono, Y; Schulten, K. Molecular Dynamics Study of Unbinding of the Avidin-Biotin Complex. Biophy. J. 1997, 72, 1568–1581. [Google Scholar]
- Florin, EL; Moy, VT; Gaub, HE. Adhesion Force between Individual Ligand-Receptor Pairs. Science 1994, 264, 415–417. [Google Scholar]
- Leach, AR. Molecular Modelling: Principles and Applications,, 2nd Ed ed; Prentice Hall; Dorset, 2001. [Google Scholar]
- Sagui, C; Darden, TA. Molecular Dynamics Simulation of Biomolecules: Long Range Electrostatic Effects. Ann. Rev. Bioph. Biomol. Struct. 1999, 28, 155–179. [Google Scholar]
- Cooke, R. Actomyosin Interaction in Striated Muscle. Phys. Rev. 1997, 77, 671–697. [Google Scholar]
- Geeves, MA; Holmes, KC. Structural Mechanism of Muscle Contraction. Annu. Rev. Biochem. 1999, 68, 687–728. [Google Scholar]
- Milligan, RA. Protein-Protein Interactions in the Rigor Actomyosin Complex. Proc. Natl. Acad. Sci. 1996, 93, 21–26. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Actomyosin(free) | Actomyosin(ATP) | Actomyosin(ADP·Pi) | Actomyosin(ADP) | |
|---|---|---|---|---|
| Fully allowed regions (%) | 62.63 | 66.84 | 64.30 | 64.31 |
| Additionally allowed regions (%) | 30.06 | 27.11 | 29.46 | 28.47 |
| Generously allowed regions (%) | 4.54 | 3.97 | 4.52 | 5.24 |
| Disallowed regions (%) | 2.77 | 2.08 | 1.72 | 1.98 |
| Actomyosin (free) | Actomyosin (ATP) | Actomyosin (ADP·Pi) | Actomyosin (ADP) | |
|---|---|---|---|---|
| S (Å2) | 833.32 | 402.08 | 543.82 | 616.09 |
| FPmax (nN) | 1.79 | 1.16 | 1.02 | 1.64 |
| Vmin (kJ/mol) | −857.03 | −224.49 | −490.67 | −422.56 |
| rVmin (nm) | 4.72 | 5.55 | 5.96 | 5.86 |
| Fmax (nN) | 0.83 | 0.27 | 0.46 | 0.60 |
| kint (pN/nm) | 6583.84 | 727.68 | 400.39 | 1139.81 |
© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aprodu, I.; Redaelli, A.; Soncini, M. Actomyosin Interaction: Mechanical and Energetic Properties in Different Nucleotide Binding States. Int. J. Mol. Sci. 2008, 9, 1927-1943. https://doi.org/10.3390/ijms9101927
Aprodu I, Redaelli A, Soncini M. Actomyosin Interaction: Mechanical and Energetic Properties in Different Nucleotide Binding States. International Journal of Molecular Sciences. 2008; 9(10):1927-1943. https://doi.org/10.3390/ijms9101927
Chicago/Turabian StyleAprodu, Iuliana, Alberto Redaelli, and Monica Soncini. 2008. "Actomyosin Interaction: Mechanical and Energetic Properties in Different Nucleotide Binding States" International Journal of Molecular Sciences 9, no. 10: 1927-1943. https://doi.org/10.3390/ijms9101927