Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain

Abstract
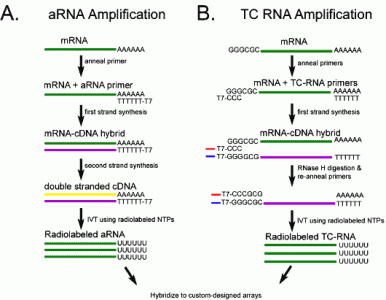
:
{kind=link}
{kind=link}
{kind=link}
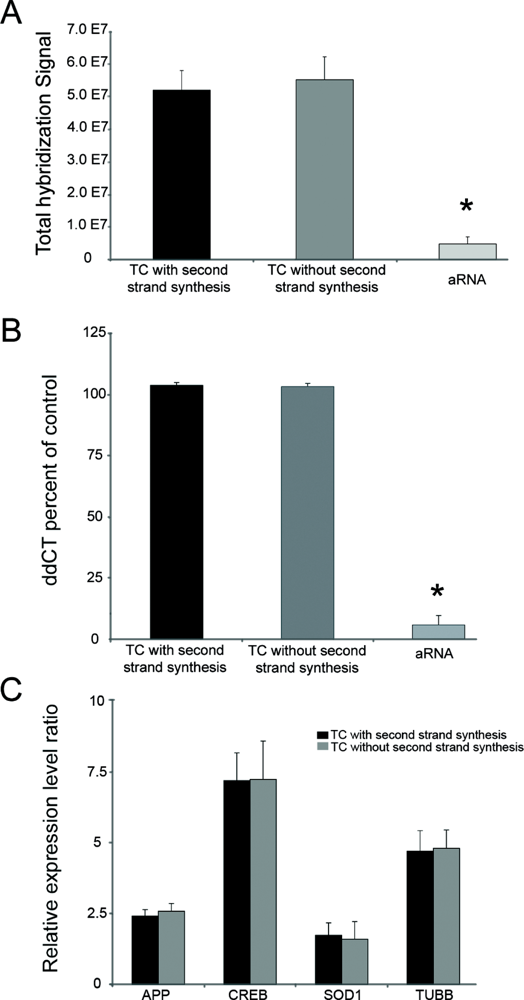{kind=link}
{kind=link}
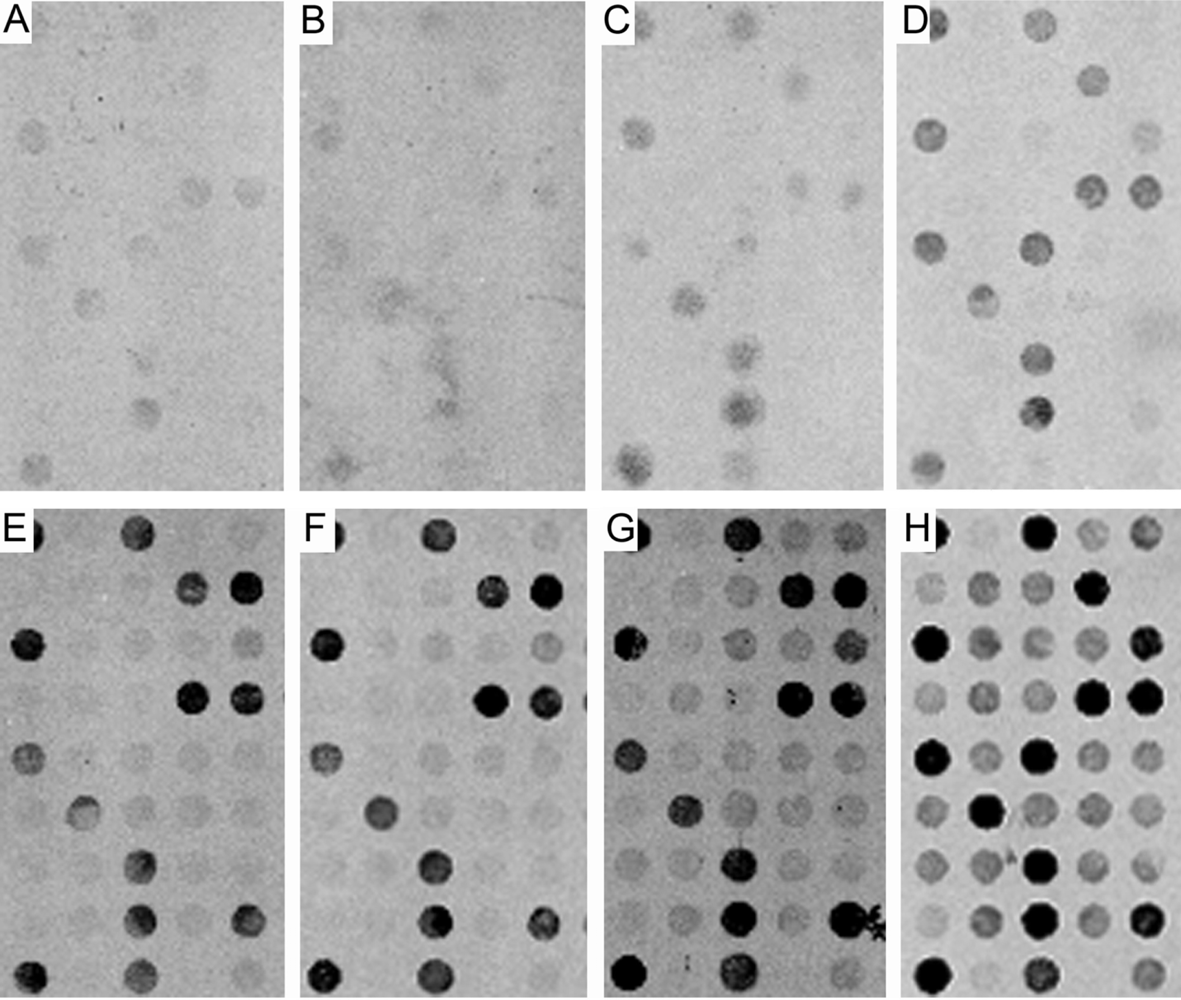{kind=link}
{kind=link}
1. Introduction
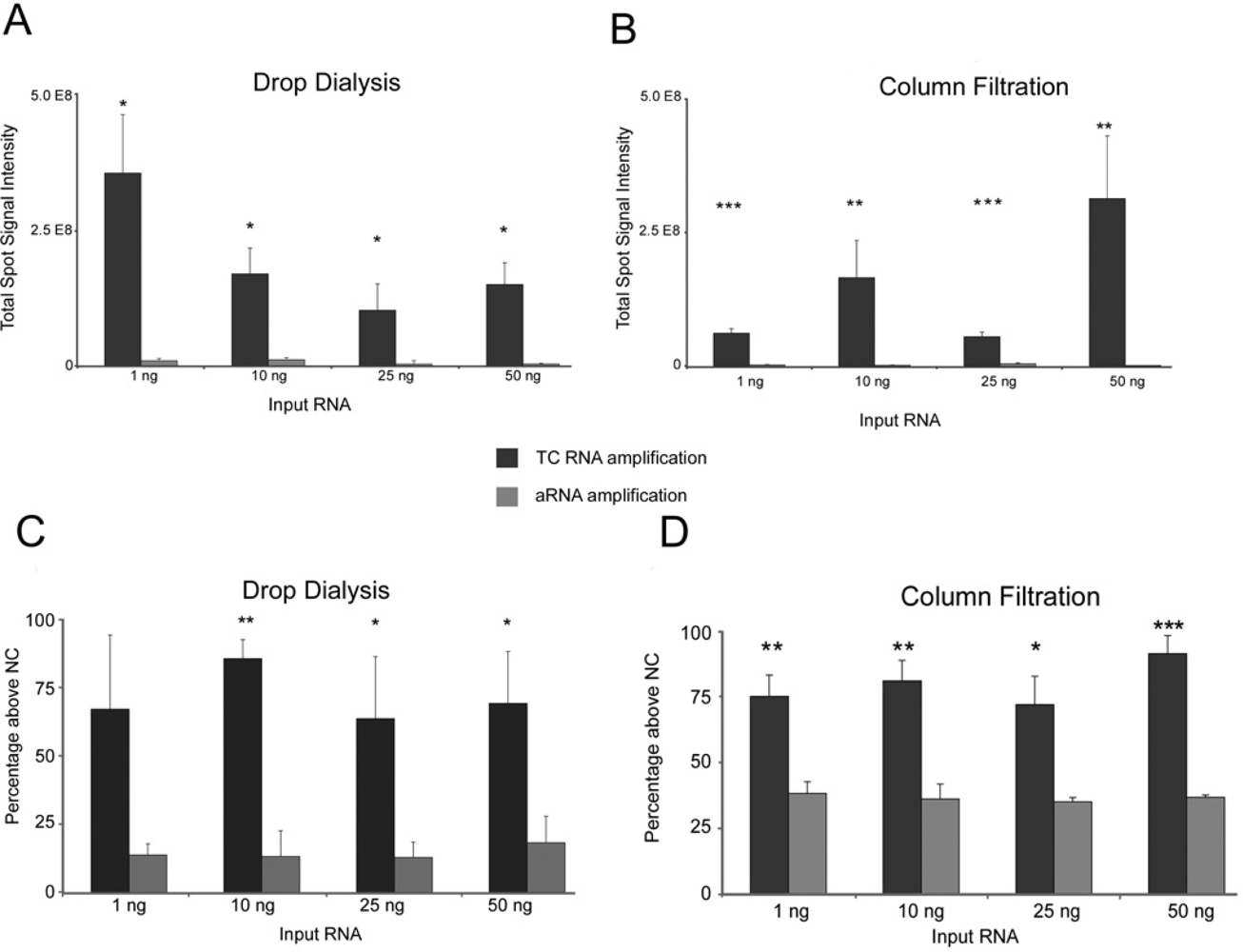
2. Results
3. Discussion
4. Materials and Methods
4.1. Tissue and RNA accession
4.2. Amplification of RNA utilizing TC RNA amplification procedure
4.3. Amplification of RNA utilizing the aRNA amplification procedure
4.4. T7 amplification and hybridization to custom-designed cDNA array platforms
4.5. Custom-designed cDNA array platforms and data analysis
4.6. Real-time quantitative PCR (qPCR)
5. Conclusions
Acknowledgments
References
- Sambrook, J; Russell, DW. Molecular Cloning: A Laboratory Manual, 3rd Ed ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Kacharmina, JE; Crino, PB; Eberwine, J. Preparation of cDNA from single cells and subcellular regions. Methods Enzymol 1999, 303, 3–18. [Google Scholar]
- Eberwine, J; Kacharmina, JE; Andrews, C; Miyashiro, K; McIntosh, T; Becker, K; Barrett, T; Hinkle, D; Dent, G; Marciano, P. mRNA expression analysis of tissue sections and single cells. J. Neurosci 2001, 21, 8310–8314. [Google Scholar]
- Ginsberg, SD. Transcriptional profiling of small samples in the central nervous system. Methods Mol. Biol 2008, 439, 147–158. [Google Scholar]
- VanGelder, R; von Zastrow, M; Yool, A; Dement, W; Barchas, J; Eberwine, J. Amplified RNA (aRNA) synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. USA 1990, 87, 1663–1667. [Google Scholar]
- Che, S; Ginsberg, SD. Amplification of transcripts using terminal continuation. Lab. Invest 2004, 84, 131–137. [Google Scholar]
- Wang, E; Miller, LD; Ohnmacht, GA; Liu, ET; Marincola, FM. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol 2000, 18, 457–459. [Google Scholar]
- Zhumabayeva, B; Diatchenko, L; Chenchik, A; Siebert, PD. Use of SMART-generated cDNA for gene expression studies in multiple human tumors. BioTechniques 2001, 30, 158–163. [Google Scholar]
- Dafforn, A; Chen, P; Deng, G; Herrler, M; Iglehart, D; Koritala, S; Lato, S; Pillarisetty, S; Purohit, R; Wang, M; Wang, SL; Kurn, N. Linear mRNA amplification from as little as 5 ng total RNA for global gene expression analysis. BioTechniques 2004, 37, 854–857. [Google Scholar]
- Kurn, N; Chen, P; Heath, JD; Kopf-Sill, A; Stephens, KM; Wang, S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin. Chem 2005, 51, 1973–1981. [Google Scholar]
- Matz, M; Shagin, D; Bogdanova, E; Britanova, O; Lukyanov, S; Diatchenko, L; Chenchik, A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res 1999, 27, 1558–1560. [Google Scholar]
- Iscove, NN; Barbara, M; Gu, M; Gibson, M; Modi, C; Winegarden, N. Representation is faithfully preserved in global cDNA amplified exponentially from sub-picogram quantities of mRNA. Nat. Biotechnol 2002, 20, 940–943. [Google Scholar]
- Xiang, CC; Chen, M; Ma, L; Phan, QN; Inman, JM; Kozhich, OA; Brownstein, MJ. A new strategy to amplify degraded RNA from small tissue samples for microarray studies. Nucleic Acids Res 2003, 31, E53. [Google Scholar]
- Goff, LA; Bowers, J; Schwalm, J; Howerton, K; Getts, RC; Hart, RP. Evaluation of sense-strand mRNA amplification by comparative quantitative PCR. BMC Genomics 2004, 5, 76. [Google Scholar]
- Counts, SE; Chen, EY; Che, S; Ikonomovic, MD; Wuua, J; Ginsberg, SD; DeKosky, ST; Mufson, EJ. Galanin fiber hypertrophy within the cholinergic nucleus basalis during the progression of Alzheimer's disease. Dement. Geriatr. Cogn. Disord 2006, 21, 205–214. [Google Scholar]
- Counts, SE; He, B; Che, S; Ikonomovic, MD; DeKosky, ST; Ginsberg, SD; Mufson, EJ. {alpha}7 Nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch. Neurol 2007, 64, 1771–1776. [Google Scholar]
- Ginsberg, SD; Che, S; Counts, SE; Mufson, EJ. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J. Neurochem 2006, 96, 1401–1408. [Google Scholar]
- Ginsberg, SD; Che, S; Wuu, J; Counts, SE; Mufson, EJ. Down regulation of trk but not p75 gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J. Neurochem 2006, 97, 475–487. [Google Scholar]
- White, MM; Sheffer, I; Teeter, J; Apostolakis, EM. Hypothalamic progesterone receptor-A mediates gonadotropin surges, self priming and receptivity in estrogen-primed female mice. J. Mol. Endocrinol 2007, 38, 35–50. [Google Scholar]
- Alldred, MJ; Che, S; Ginsbberg, SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J. Neurosci. Meth. in press..
- Ginsberg, SD; Che, S. RNA amplification in brain tissues. Neurochem. Res 2002, 27, 981–992. [Google Scholar]
- Hemby, SE; Ginsberg, SD; Brunk, B; Arnold, SE; Trojanowski, JQ; Eberwine, JH. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch. Gen. Psychiat 2002, 59, 631–640. [Google Scholar]
- Zhao, X; Lein, ES; He, A; Smith, SC; Aston, C; Gage, FH. Transcriptional profiling reveals strict boundaries between hippocampal subregions. J. Comp. Neurol 2001, 441, 187–196. [Google Scholar]
- Vincent, VA; DeVoss, JJ; Ryan, HS; Murphy, GM, Jr. Analysis of neuronal gene expression with laser capture microdissection. J. Neurosci. Res 2002, 69, 578–586. [Google Scholar]
- Lu, L; Neff, F; Dun, Z; Hemmer, B; Oertel, WH; Schlegel, J; Hartmann, A. Gene expression profiles derived from single cells in human postmortem brain. Brain Res. Brain Res. Protoc 2004, 13, 18–25. [Google Scholar]
- Hemby, SE; Trojanowski, JQ; Ginsberg, SD. Neuron-specific age-related decreases in dopamine receptor subtype mRNAs. J. Comp. Neurol 2003, 456, 176–183. [Google Scholar]
- Ginsberg, SD; Che, S. Expression profile analysis within the human hippocampus: Comparison of CA1 and CA3 pyramidal neurons. J. Comp. Neurol 2005, 487, 107–118. [Google Scholar]
- Ginsberg, SD; Che, S; Counts, SE; Mufson, EJ. Single cell gene expression profiling in Alzheimer's disease. NeuroRx 2006, 3, 302–318. [Google Scholar]
- Kamme, F; Salunga, R; Yu, J; Tran, DT; Zhu, J; Luo, L; Bittner, A; Guo, H-Q; Miller, N; Wan, J; Erlander, M. Single-cell microarray analysis in hippocampus CA1: Demonstration and validation of cellular heterogeneity. J. Neurosci 2003, 23, 3607–3615. [Google Scholar]
- Zhu, B; Xu, F; Baba, Y. An evaluation of linear RNA amplification in cDNA microarray gene expression analysis. Mol. Genet. Metab 2006, 87, 71–79. [Google Scholar]
- Penland, SK; Keku, TO; Torrice, C; He, X; Krishnamurthy, J; Hoadley, KA; Woosley, JT; Thomas, NE; Perou, CM; Sandler, RS; Sharpless, NE. RNA expression analysis of formalin-fixed paraffin-embedded tumors. Lab. Invest 2007, 87, 383–391. [Google Scholar]
- Ding, Y; Xu, L; Chen, S; Jovanovic, BD; Helenowski, IB; Kelly, DL; Catalona, WJ; Yang, XJ; Pins, M; Ananthanarayanan, V; Bergan, RC. Characterization of a method for profiling gene expression in cells recovered from intact human prostate tissue using RNA linear amplification. Prostate Cancer Prostatic Dis 2006, 9, 379–391. [Google Scholar]
- Baugh, LR; Hill, AA; Brown, EL; Hunter, CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res 2001, 29, E29. [Google Scholar]
- Dixon, AK; Lee, K; Richardson, PJ; Bell, MI; Skynner, MJ. Single cell expression analysis - pharmacogenomic potential. Pharmacogenomics 2002, 3, 809–822. [Google Scholar]
- Marko, NF; Frank, B; Quackenbush, J; Lee, NH. A robust method for the amplification of RNA in the sense orientation. BMC Genomics 2005, 6, 27. [Google Scholar]
- Ginsberg, S. D. RNA amplification strategies for small sample populations. Methods 2005, 37, 229–237. [Google Scholar]





© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alldred, M.J.; Che, S.; Ginsberg, S.D. Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain. Int. J. Mol. Sci. 2008, 9, 2091-2104. https://doi.org/10.3390/ijms9112091
Alldred MJ, Che S, Ginsberg SD. Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain. International Journal of Molecular Sciences. 2008; 9(11):2091-2104. https://doi.org/10.3390/ijms9112091
Chicago/Turabian StyleAlldred, Melissa J., Shaoli Che, and Stephen D. Ginsberg. 2008. "Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain" International Journal of Molecular Sciences 9, no. 11: 2091-2104. https://doi.org/10.3390/ijms9112091