Contribution of Natural Inhibitors to the Understanding of the PI3K/PDK1/PKB Pathway in the Insulin-mediated Intracellular Signaling Cascade


Abstract
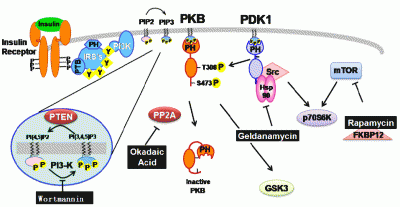
:
{kind=link}
{kind=link}
{kind=link}
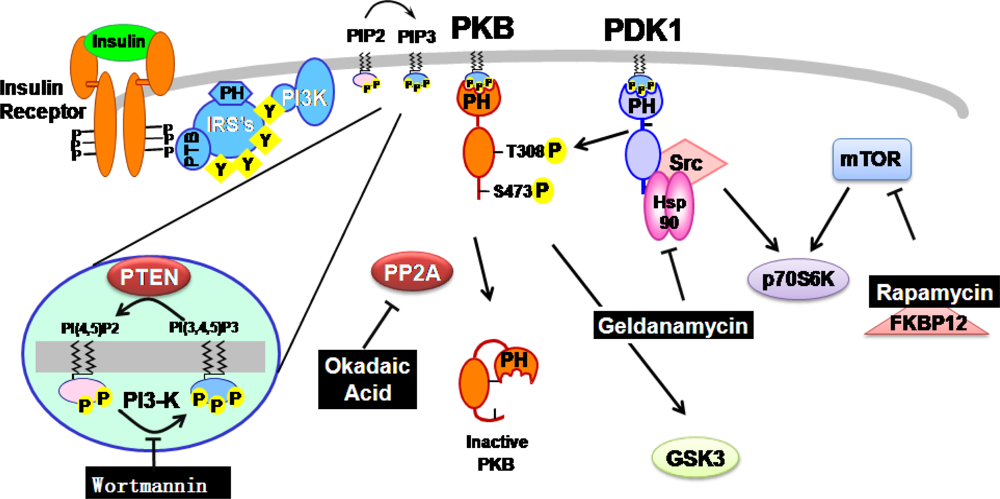{kind=link}
1. Introduction
2. Natural inhibitors targeting PI3K/PDK1/PKB pathway in insulin-mediated signaling events
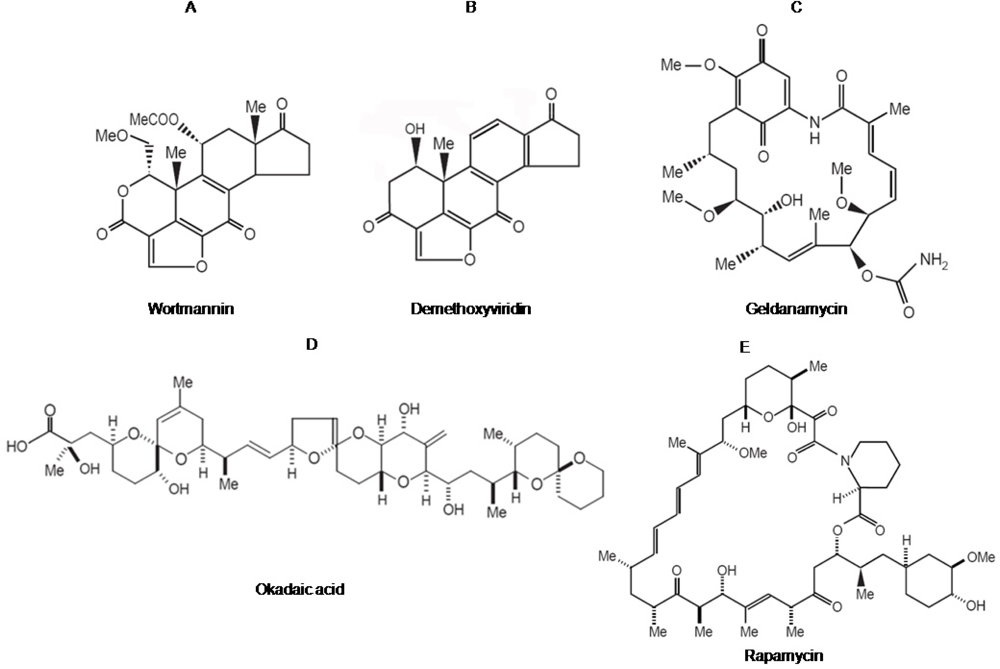
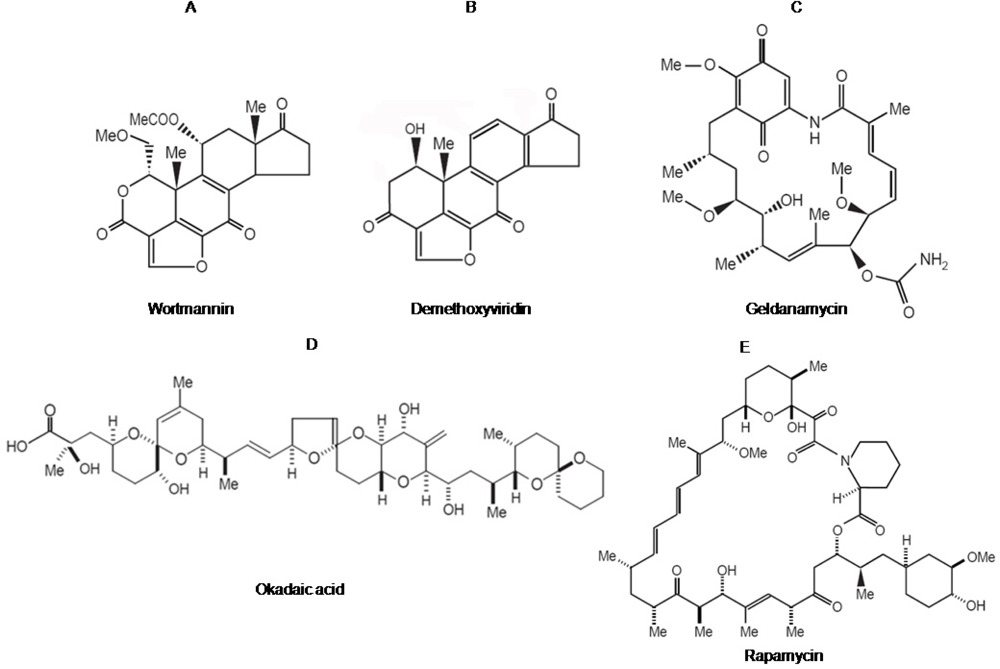
2.1. Wortmannin: An inhibitor of PI-3-kinase family members
2.2. Geldanamycin: An inhibitor of Hsp90 and of Hsp90-dependent-signalling components
2.3. Okadaic acid : An inhibitor of serine/threonine protein phosphatase
2.4. Rapamycin: an inhibitor of mTOR-dependent signaling pathways
3. Conclusions
Acknowledgments
References and Notes
- Foster, DW; McGarry, JD. The metabolic derangements and treatment of diabetic ketoacidosis. N. Engl. J. Med 1983, 309, 159–169. [Google Scholar]
- Coccheri, S. Approaches to prevention of cardiovascular complications and events in diabetes mellitus. Drugs 2007, 67, 997–1026. [Google Scholar]
- Bretzel, RG; Eckhard, M; Brendel, MD. Pancreatic islet and stem cell transplantation: New strategies in cell therapy of diabetes mellitus. Panminerva Med 2004, 46, 25–42. [Google Scholar]
- Kahn, BB. Type 2 diabetes: When insulin secretion fails to compensate for insulin resistance. Cell 1998, 92, 593–596. [Google Scholar]
- Saltiel, AR; Pessin, JE. Insulin signaling pathways in time and space. Trends Cell. Biol 2002, 12, 65–71. [Google Scholar]
- Taylor, SI. Deconstructing type 2 diabetes. Cell 1999, 97, 9–12. [Google Scholar]
- Moller, DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature 2001, 414, 821–827. [Google Scholar]
- Kahan, BD. Sirolimus-based immunosuppression: present state of the art. J. Nephrol 2004, 17(Suppl 8), S32–39. [Google Scholar]
- Thompson, EJ; MacGowan, J; Young, MR; Colburn, N; Bowden, GT. A dominant negative c-jun specifically blocks okadaic acid-induced skin tumor promotion. Cancer Res 2002, 62, 3044–3047. [Google Scholar]
- Whitesell, L; Mimnaugh, EG; De Costa, B; Myers, CE; Neckers, LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar]
- Ui, M; Okada, T; Hazeki, K; Hazeki, O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci 1995, 20, 303–307. [Google Scholar]
- Martinez, D; Vermeulen, M; Trevani, A; Ceballos, A; Sabatte, J; Gamberale, R; Alvarez, ME; Salamone, G; Tanos, T; Coso, OA; Geffner, J. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J. Immunol 2006, 176, 1163–1171. [Google Scholar]
- Pandey, V; Mihara, S; Fensome-Green, A; Bolsover, S; Cockcroft, S. Monomeric IgE stimulates NFAT translocation into the nucleus, a rise in cytosol Ca2+, degranulation, and membrane ruffling in the cultured rat basophilic leukemia-2H3 mast cell line. J. Immunol 2004, 172, 4048–4058. [Google Scholar]
- Lee, JY; Kim, JY; Lee, YG; Shin, WC; Chun, T; Rhee, MH; Cho, JY. Hydroquinone, a reactive metabolite of benzene, reduces macrophage-mediated immune responses. Mol. Cells 2007, 23, 198–206. [Google Scholar]
- Falasca, M; Maffucci, T. Role of class II phosphoinositide 3-kinase in cell signalling. Biochem. Soc. Trans 2007, 35, 211–214. [Google Scholar]
- Carnero, A; Blanco-Aparicio, C; Renner, O; Link, W; Leal, JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar]
- Cifuentes, M; Rojas, CV. Antilipolytic effect of calcium-sensing receptor in human adipocytes. Mol. Cell. Biochem 2008, 319, 17–21. [Google Scholar]
- Li, HB; Ge, YK; Zheng, XX; Zhang, L. Salidroside stimulated glucose uptake in skeletal muscle cells by activating AMP-activated protein kinase. Eur. J. Pharmacol 2008, 588, 165–169. [Google Scholar]
- Kanazawa, H; Ohsawa, K; Sasaki, Y; Kohsaka, S; Imai, Y. Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma -dependent pathway. J. Biol. Chem 2002, 277, 20026–20032. [Google Scholar]
- Shimaya, A; Kovacina, KS; Roth, RA. On the mechanism for neomycin reversal of wortmannin inhibition of insulin stimulation of glucose uptake. J. Biol. Chem 2004, 279, 55277–55282. [Google Scholar]
- Tang, X; Wang, L; Proud, CG; Downes, CP. Muscarinic receptor-mediated activation of p70 S6 kinase 1 (S6K1) in 1321N1 astrocytoma cells: permissive role of phosphoinositide 3-kinase. Biochem. J 2003, 374, 137–143. [Google Scholar]
- Hinault, C; Mothe-Satney, I; Gautier, N; Van Obberghen, E. Amino acids require glucose to enhance, through phosphoinositide-dependent protein kinase 1, the insulin-activated protein kinase B cascade in insulin-resistant rat adipocytes. Diabetologia 2006, 49, 1017–1026. [Google Scholar]
- Jensen, J; Brennesvik, EO; Lai, YC; Shepherd, PR. GSK-3beta regulation in skeletal muscles by adrenaline and insulin: evidence that PKA and PKB regulate different pools of GSK-3. Cell. Signal 2007, 19, 204–210. [Google Scholar]
- Hennersdorf, F; Florian, S; Jakob, A; Baumgartner, K; Sonneck, K; Nordheim, A; Biedermann, T; Valent, P; Buhring, HJ. Identification of CD13, CD107a, and CD164 as novel basophil-activation markers and dissection of two response patterns in time kinetics of IgE-dependent upregulation. Cell Res 2005, 15, 325–335. [Google Scholar]
- Arndt, PG; Suzuki, N; Avdi, NJ; Malcolm, KC; Worthen, GS. Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-Kinase and Syk-mediated pathways. J. Biol. Chem 2004, 279, 10883–10891. [Google Scholar]
- Okada, T; Sakuma, L; Fukui, Y; Hazeki, O; Ui, M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. J. Biol. Chem 1994, 269, 3563–3567. [Google Scholar]
- Wymann, MP; Bulgarelli-Leva, G; Zvelebil, MJ; Pirola, L; Vanhaesebroeck, B; Waterfield, MD; Panayotou, G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol. Cell. Biol 1996, 16, 1722–1733. [Google Scholar]
- Meyers, R; Cantley, LC. Cloning and characterization of a wortmannin-sensitive human phosphatidylinositol 4-kinase. J. Biol. Chem 1997, 272, 4384–4390. [Google Scholar]
- Woscholski, R; Kodaki, T; McKinnon, M; Waterfield, MD; Parker, PJ. A comparison of demethoxyviridin and wortmannin as inhibitors of phosphatidylinositol 3-kinase. FEBS Lett 1994, 342, 109–114. [Google Scholar]
- Stack, JH; Emr, SD. Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J. Biol. Chem 1994, 269, 31552–31562. [Google Scholar]
- Cutler, NS; Heitman, J; Cardenas, ME. STT4 is an essential phosphatidylinositol 4-kinase that is a target of wortmannin in Saccharomyces cerevisiae. J. Biol. Chem 1997, 272, 27671–27677. [Google Scholar]
- Brunn, GJ; Williams, J; Sabers, C; Wiederrecht, G; Lawrence, JC, Jr; Abraham, RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J 1996, 15, 5256–5267. [Google Scholar]
- Alarcon, CM; Heitman, J; Cardenas, ME. Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Mol. Biol. Cell 1999, 10, 2531–2546. [Google Scholar]
- Sarkaria, JN; Tibbetts, RS; Busby, EC; Kennedy, AP; Hill, DE; Abraham, RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 1998, 58, 4375–4382. [Google Scholar]
- Hartley, KO; Gell, D; Smith, GC; Zhang, H; Divecha, N; Connelly, MA; Admon, A; Lees-Miller, SP; Anderson, CW; Jackson, SP. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995, 82, 849–856. [Google Scholar]
- Stravopodis, DJ; Margaritis, LH; Voutsinas, GE. Drug-mediated targeted disruption of multiple protein activities through functional inhibition of the Hsp90 chaperone complex. Curr. Med. Chem 2007, 14, 3122–3138. [Google Scholar]
- Prodromou, C; Pearl, LH. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar]
- Roe, SM; Prodromou, C; O'Brien, R; Ladbury, JE; Piper, PW; Pearl, LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem 1999, 42, 260–266. [Google Scholar]
- Sato, S; Fujita, N; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. U S A 2000, 97, 10832–10837. [Google Scholar]
- Wei, Q; Xia, Y. Roles of 3-phosphoinositide-dependent kinase 1 in the regulation of endothelial nitric-oxide synthase phosphorylation and function by heat shock protein 90. J. Biol. Chem 2005, 280, 18081–18086. [Google Scholar]
- Fontana, J; Fulton, D; Chen, Y; Fairchild, TA; McCabe, TJ; Fujita, N; Tsuruo, T; Sessa, WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ. Res 2002, 90, 866–873. [Google Scholar]
- Yun, BG; Matts, RL. Hsp90 functions to balance the phosphorylation state of Akt during C2C12 myoblast differentiation. Cell Signal 2005, 17, 1477–1485. [Google Scholar]
- Fujita, N; Tsuruo, T. Survival-signaling pathway as a promising target for cancer chemotherapy. Cancer Chemother. Pharmacol 2003, 52(Suppl 1), S24–28. [Google Scholar]
- Supko, JG; Hickman, RL; Grever, MR; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol 1995, 36, 305–315. [Google Scholar]
- Schnur, RC; Corman, ML; Gallaschun, RJ; Cooper, BA; Dee, MF; Doty, JL; Muzzi, ML; Moyer, JD; DiOrio, CI; Barbacci, EG; et al. Inhibition of the oncogene product p185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J. Med. Chem 1995, 38, 3806–3812. [Google Scholar]
- Scheuer, PJ. Marine natural products. Diversity in molecular structure and bioactivity. Adv. Exp. Med. Biol 1996, 391, 1–8. [Google Scholar]
- Suganuma, M; Fujiki, H; Suguri, H; Yoshizawa, S; Hirota, M; Nakayasu, M; Ojika, M; Wakamatsu, K; Yamada, K; Sugimura, T. Okadaic acid: an additional non-phorbol-12-tetradecanoate-13-acetate-type tumor promoter. Proc. Natl. Acad. Sci. USA 1988, 85, 1768–1771. [Google Scholar]
- Rami, BG; Chin, LS; Lazio, BE; Singh, SK. Okadaic-acid-induced apoptosis in malignant glioma cells. Neurosurg. Focus 2003, 14, e4. [Google Scholar]
- Dounay, AB; Forsyth, CJ. Okadaic acid: the archetypal serine/threonine protein phosphatase inhibitor. Curr. Med. Chem 2002, 9, 1939–1980. [Google Scholar]
- McCluskey, A; Sim, AT; Sakoff, JA. Serine-threonine protein phosphatase inhibitors: development of potential therapeutic strategies. J. Med. Chem 2002, 45, 1151–1175. [Google Scholar]
- Matsuzawa, S; Suzuki, T; Suzuki, M; Matsuda, A; Kawamura, T; Mizuno, Y; Kikuchi, K. Thyrsiferyl 23-acetate is a novel specific inhibitor of protein phosphatase PP2A. FEBS Lett 1994, 356, 272–274. [Google Scholar]
- Li, YM; Casida, JE. Cantharidin-binding protein: identification as protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1992, 89, 11867–11870. [Google Scholar]
- Bryant, NJ; Govers, R; James, DE. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell. Biol 2002, 3, 267–277. [Google Scholar]
- Dickey, RW; Bobzin, SC; Faulkner, DJ; Bencsath, FA; Andrzejewski, D. Identification of okadaic acid from a Caribbean dinoflagellate, Prorocentrum concavum. Toxicon 1990, 28, 371–377. [Google Scholar]
- Dounay, AB; Urbanek, RA; Sabes, SF; Forsyth, CJ. Total Synthesis of the Marine Natural Product 7-Deoxy-okadaic Acid: A Potent Inhibitor of Serine/Threonine-Specific Protein Phosphatases. Angew. Chem. Int. Ed. Engl 1999, 38, 2258–2262. [Google Scholar]
- Dounay, AB; Urbanek, RA; Frydrychowski, VA; Forsyth, CJ. Expedient access to the okadaic acid architecture: a novel synthesis of the C1–C27 domain. J. Org. Chem 2001, 66, 925–938. [Google Scholar]
- Kita, A; Matsunaga, S; Takai, A; Kataiwa, H; Wakimoto, T; Fusetani, N; Isobe, M; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar]
- Cardenas, ME; Cutler, NS; Lorenz, MC; Di Como, CJ; Heitman, J. The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev 1999, 13, 3271–3279. [Google Scholar]
- Sormani, R; Yao, L; Menand, B; Ennar, N; Lecampion, C; Meyer, C; Robaglia, C. Saccharomyces cerevisiae FKBP12 binds Arabidopsis thaliana TOR and its expression in plants leads to rapamycin susceptibility. BMC Plant Biol 2007, 7, 26. [Google Scholar]
- Krummrei, U; Baulieu, EE; Chambraud, B. The FKBP-associated protein FAP48 is an antiproliferative molecule and a player in T cell activation that increases IL2 synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2444–2449. [Google Scholar]
- Crespo, JL; Hall, MN. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev 2002, 66, 579–591. [Google Scholar]
- Helliwell, SB; Wagner, P; Kunz, J; Deuter-Reinhard, M; Henriquez, R; Hall, MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol. Biol. Cell 1994, 5, 105–118. [Google Scholar]
- Sabers, CJ; Martin, MM; Brunn, GJ; Williams, JM; Dumont, FJ; Wiederrecht, G; Abraham, RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem 1995, 270, 815–822. [Google Scholar]
- Dames, SA. A fast and simple method to prepare the FKBP-rapamycin binding domain of human target of rapamycin for NMR binding assays. Protein Expr. Purif 2008, 59, 31–37. [Google Scholar]
- Lorenz, MC; Heitman, J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem 1995, 270, 27531–27537. [Google Scholar]
- Choi, J; Chen, J; Schreiber, SL; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar]
- Joo, EK; Broxmeyer, HE; Kwon, HJ; Kang, HB; Kim, JS; Lim, JS; Choe, YK; Choe, IS; Myung, PK; Lee, Y. Enhancement of cell survival by stromal cell-derived factor-1/CXCL12 involves activation of CREB and induction of Mcl-1 and c-Fos in factor-dependent human cell line MO7e. Stem Cells Dev 2004, 13, 563–570. [Google Scholar]
- Lawrence, JC; Lin, TA; McMahon, LP; Choi, KM. Modulation of the protein kinase activity of mTOR. Curr. Top. Microbiol. Immunol 2004, 279, 199–213. [Google Scholar]
- McMahon, LP; Yue, W; Santen, RJ; Lawrence, JC, Jr. Farnesylthiosalicylic acid inhibits mammalian target of rapamycin (mTOR) activity both in cells and in vitro by promoting dissociation of the mTOR-raptor complex. Mol. Endocrinol 2005, 19, 175–183. [Google Scholar]
- Lee, HW; Nam, KO; Park, SJ; Kwon, BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur. J. Immunol 2003, 33, 2133–2141. [Google Scholar]
- Eto, I. Nutritional and chemopreventive anti-cancer agents up-regulate expression of p27Kip1, a cyclin-dependent kinase inhibitor, in mouse JB6 epidermal and human MCF7, MDA-MB-321 and AU565 breast cancer cells. Cancer Cell Int 2006, 6, 20. [Google Scholar]

© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cho, J.Y.; Park, J. Contribution of Natural Inhibitors to the Understanding of the PI3K/PDK1/PKB Pathway in the Insulin-mediated Intracellular Signaling Cascade. Int. J. Mol. Sci. 2008, 9, 2217-2230. https://doi.org/10.3390/ijms9112217
Cho JY, Park J. Contribution of Natural Inhibitors to the Understanding of the PI3K/PDK1/PKB Pathway in the Insulin-mediated Intracellular Signaling Cascade. International Journal of Molecular Sciences. 2008; 9(11):2217-2230. https://doi.org/10.3390/ijms9112217
Chicago/Turabian StyleCho, Jae Youl, and Jongsun Park. 2008. "Contribution of Natural Inhibitors to the Understanding of the PI3K/PDK1/PKB Pathway in the Insulin-mediated Intracellular Signaling Cascade" International Journal of Molecular Sciences 9, no. 11: 2217-2230. https://doi.org/10.3390/ijms9112217