Recognition Properties and Competitive Assays of a Dual Dopamine/Serotonin Selective Molecularly Imprinted Polymer

Abstract
:1. Introduction
2. Results and Discussion
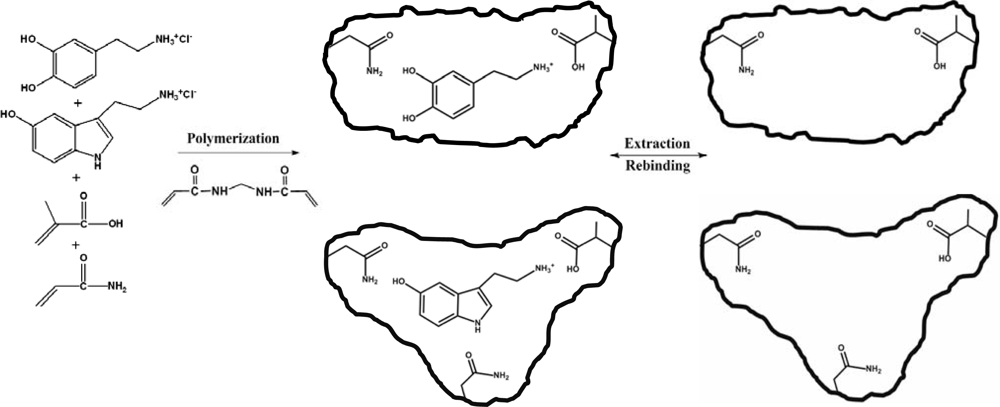
2.1. Synthesis and characterisation of DS-MIP
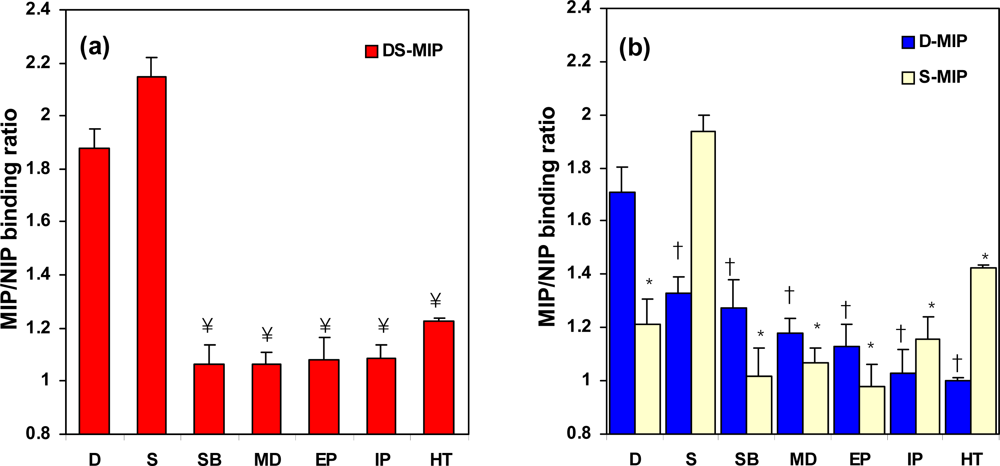
2.2. Template recognition by DS-MIP
2.3. Binding characteristics of DS-MIP
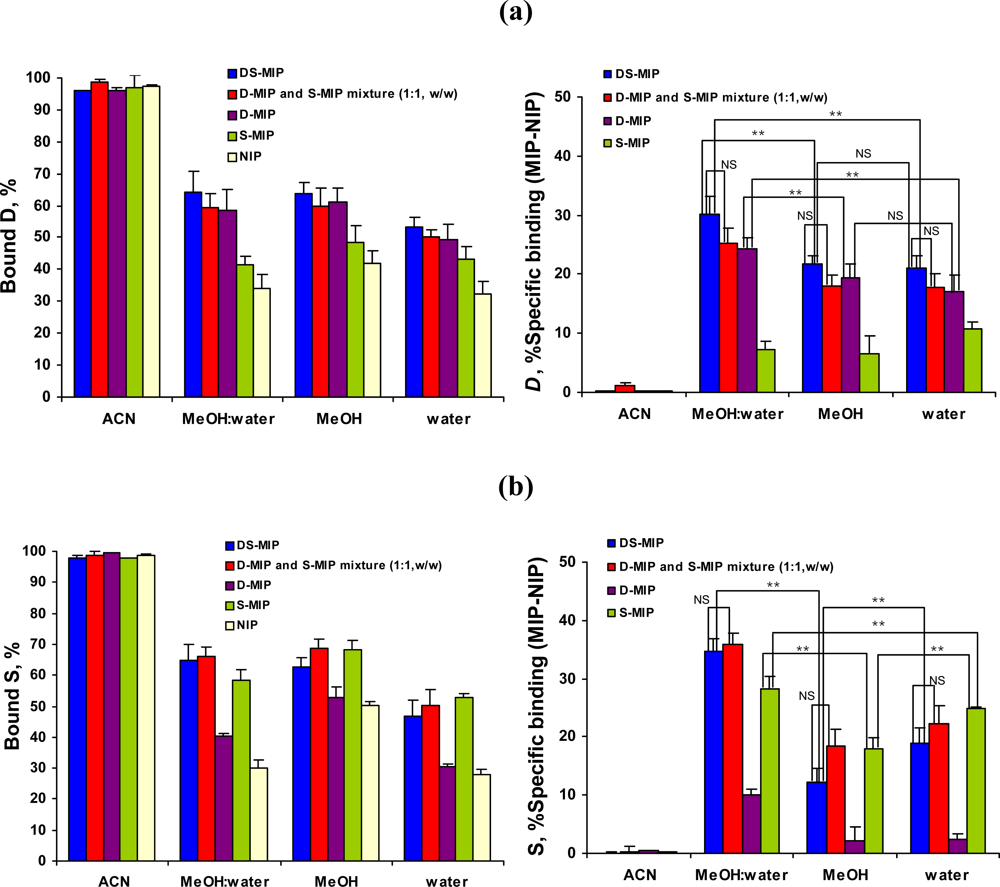
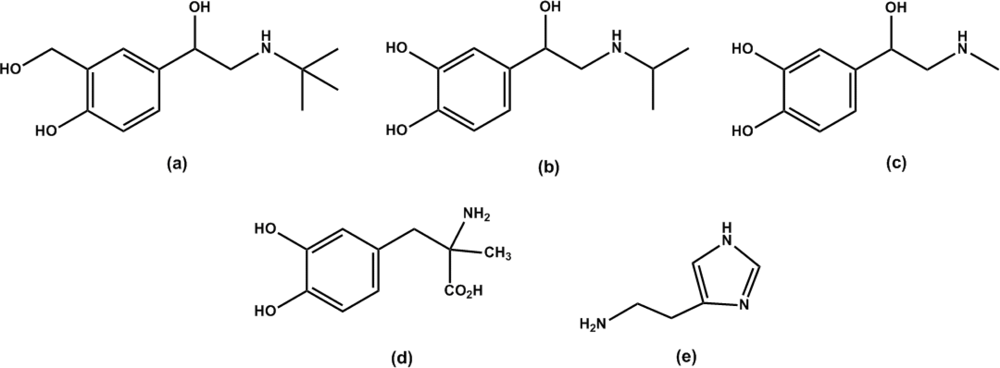
2.4. The specificity of DS-MIP
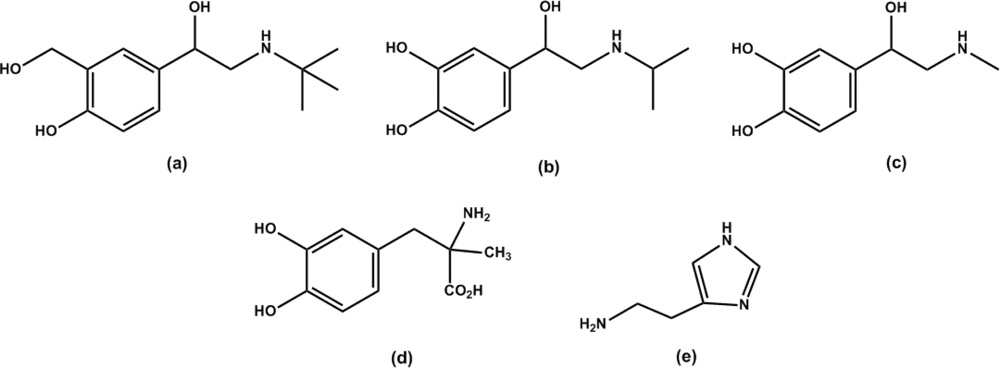
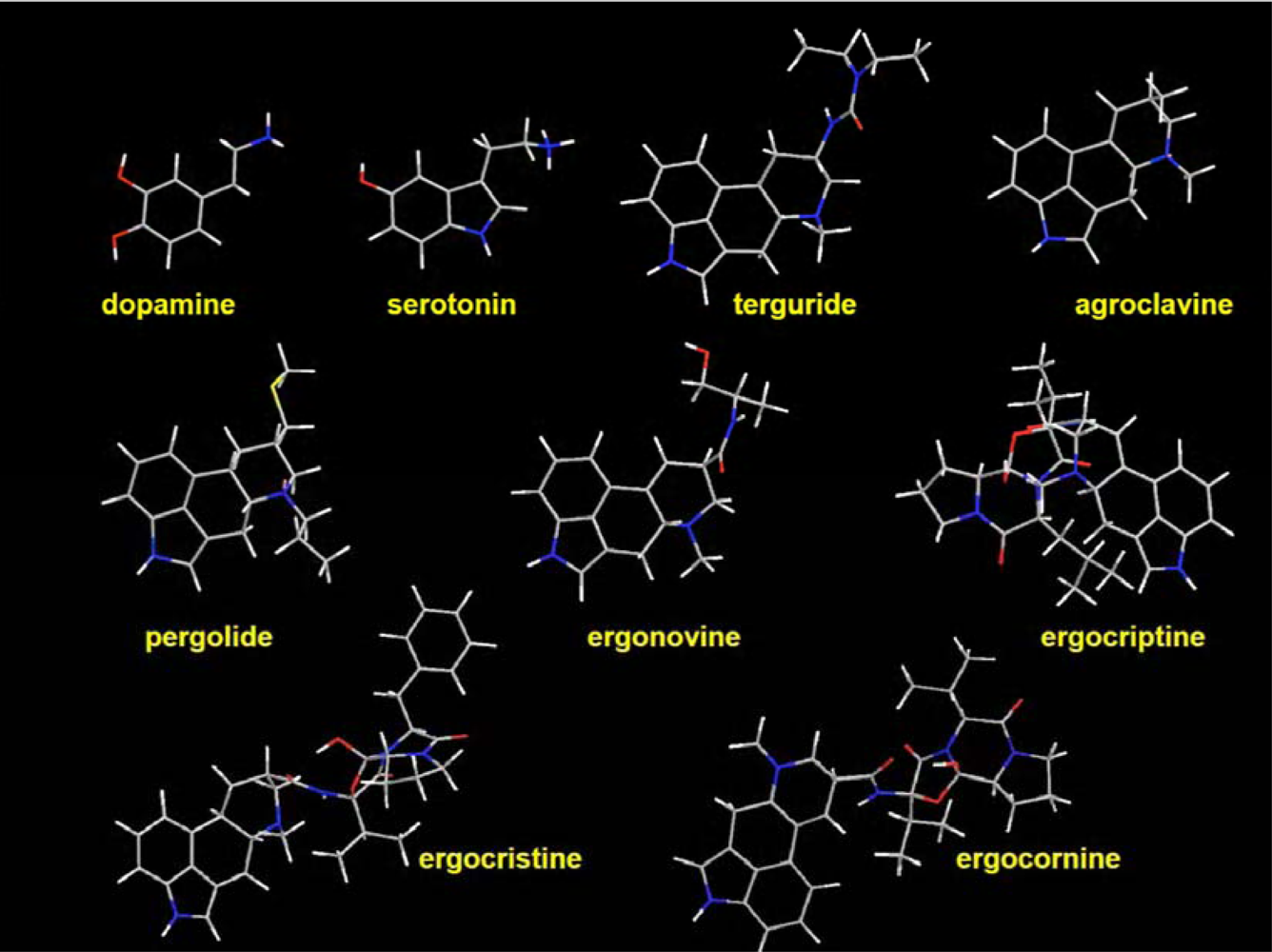
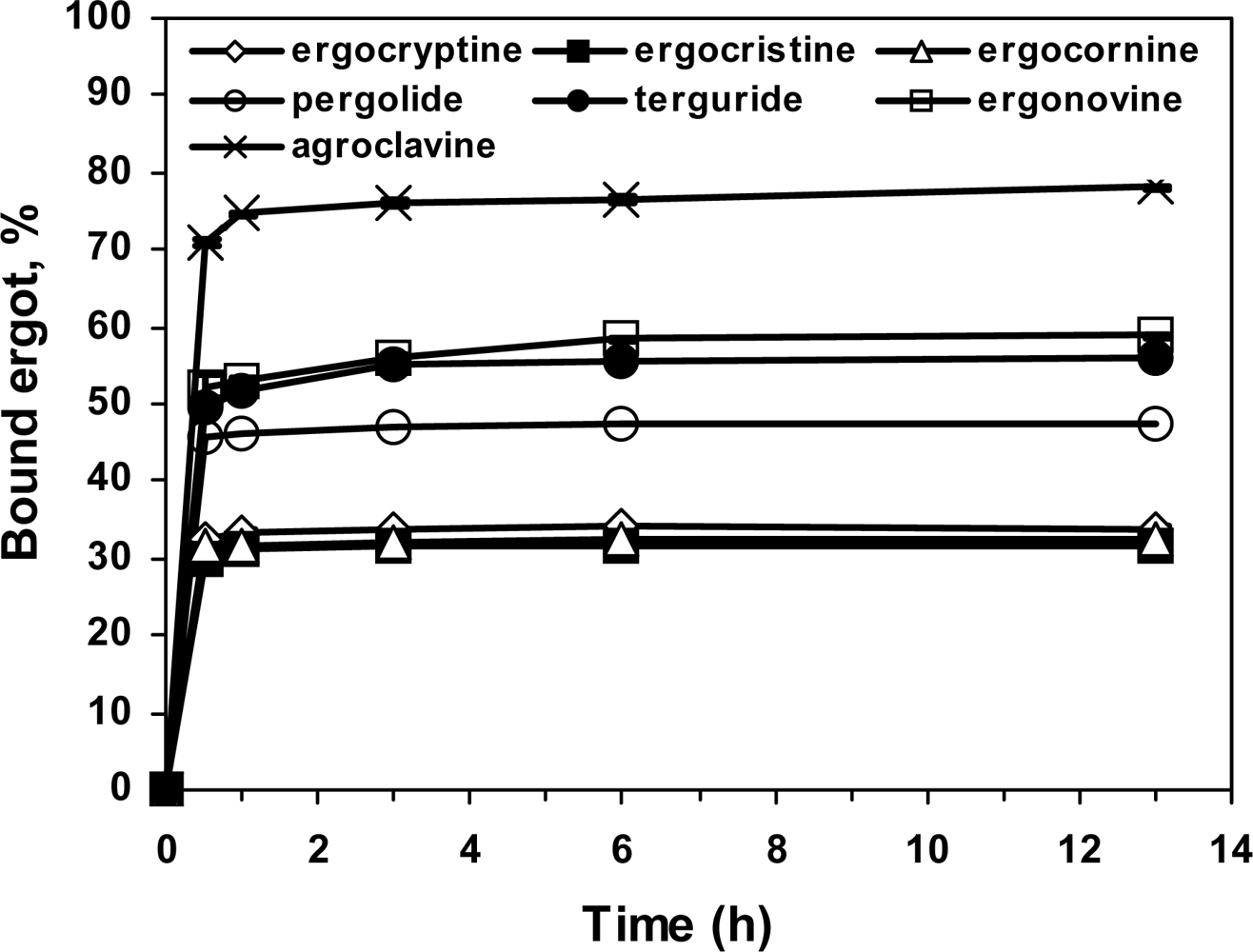
2.5. Specificity of the ergot derivatives towards DS-MIP
2.6. Affinity of the ergot derivatives towards DS-MIP
3. Experimental Section
3.1. Chemicals
3.2. Apparatus
3.3. Investigation of template/functional monomer complexation by 1H-NMR spectroscopy
3.4. Polymer synthesis
3.5. Determination of the selective recognition ability of DS-MIP
3.6. Determination of the binding characteristics of DS-MIP
3.7. Determination of the specificity of DS-MIP
3.8. Determination of the binding-reactivity of the ergot derivatives
3.9. Competitive ligand binding assays for the tested ergot derivatives
3.10. Determination of the binding affinities of the ergot derivatives to the natural receptor
3.11. Statistical analysis
4. Conclusions
Acknowledgments
References and Notes
- Kempe, M; Mosbach, K. Receptor binding mimetics: A novel molecularly imprinted polymer. Tetrahedron Lett 1995, 36, 3563–3566. [Google Scholar]
- Ramström, O; Yu, C; Mosbach, K. Chiral recognition in adrenergic receptor binding mimics prepared by molecular imprinting. J. Mol. Recog 1996, 9, 691–696. [Google Scholar]
- Alexander, C; Smith, CR; Whitcombe, MJ; Vulfson, EN. Imprinted polymers as protecting group for regioselective modification of polyfunctional substrates. J. Am. Chem. Soc 1999, 121, 6640–6651. [Google Scholar]
- Whitcombe, MJ; Vulfson, EN. Imprinted polymer. Adv. Mater 2001, 13, 467–478. [Google Scholar]
- Mayes, AG; Whitcombe, MJ. Synthetic strategies for the generation of molecularly imprinted organic polymers. Adv. Drug Deliver. Rev 2005, 57, 1742–1778. [Google Scholar]
- Alexander, C; Andersson, HS; Andersson, LI; Ansell, RJ; Kirsch, N; Nicholls, IA; O’Mahony, J; Whitcombe, MJ. Molecular imprinting science and technology: A survey of the literature for the years up to and including 2003. J. Mol. Recog 2006, 19, 106–180. [Google Scholar]
- Vlatakis, G; Andersson, LI; Muller, R; Mosbach, K. Drug assay using antibody mimics made by molecular imprinting. Nature 1993, 361, 645–647. [Google Scholar]
- Lavignac, N; Allender, CJ; Brain, KR. Current status of molecularly imprinted polymers as alternatives to antibodies in sorbent assays. Anal. Chim. Acta 2004, 510, 139–145. [Google Scholar] [Green Version]
- Piletsky, SA; Piletskaya, EV; Elskaya, AV; Levi, R; Yano, K; Karube, I. Optical detection system for triazine based on molecularly-imprinted polymers. Anal. Lett 1997, 30, 445–455. [Google Scholar]
- Haupt, K; Mayes, AG; Mosbach, K. Herbicide assay using an imprinted polymer based system analogous to competitive fluoroimmunoassays. Anal. Chem 1998, 70, 3936–3939. [Google Scholar]
- Masson, J; Sagne, C; Hamon, M; El. Mestikawy, S. Neurotransmitter transporters in the central nervous system. Pharmacol. Rev 1999, 51, 439–464. [Google Scholar]
- Amara, SG; Sonders, MS. Neurotransmitter transporters as molecular targets for addictive drugs. Drug Alcohol Depend 1998, 51, 87–96. [Google Scholar]
- Rothman, RB; Blough, BE; Baumann, MH. Dual dopamine/serotonin releasers as potential medications for stimulant and alcohol addictions. AAPS Journal 2007, 9, E1–E10. [Google Scholar]
- Liesbeth, ABS; Mark, SK; Adrian, N-T. Effects of novel antipsychotics with mixed D2 antagonist/5-HT1A agonist properties on PCP-induced social interaction deficits in the rat. Neuropharmacology 2005, 49, 996–1006. [Google Scholar]
- Murakami, Y; Lehn, J-M (Eds.) Atwood, J.L., Davis, J.E.D., Macnicol, D.D., Vögtle, F., Executive Eds.; Supramolecular Reactivity and Transport: Bioorganic Systems. Comprehensive Supramolecular Chemistry; Elsevier: Amsterdam, 1996; Volume 3, pp. 1–40.
- Hollenberg, MD. Neurotransmitter Receptor Binding, 2nd Ed; Yamamura, HJ, Enna, SJ, Kuhar, MJ, Eds.; Raven Press: New York, 1985; pp. 1–40. [Google Scholar]
- Matsui, J; Sodeyama, T; Tamaki, K; Sukimoto, N. Molecularly-imprinted polymeric gates selective for predetermined chemical input species. Chem. Commun 2006, 3217–3219. [Google Scholar]
- Sreenivasan, K. Molecularly imprinted polyacrylic acid containing multiple recognition sites for steroids. J. Appl. Polym. Sci 2001, 82, 889–893. [Google Scholar]
- Suedee, R; Srichana, T; Chuchome, T; Kongmark, U. Use of molecularly imprinted polymers from a mixture of tetracycline and its degradation products to produce affinity membranes for the removal of tetracycline from water. J. Chromatogr. B 2004, 811, 191–200. [Google Scholar]
- Gilman, AG; Goodman, LS. The Pharmacological basis of therapeutic; Gilman, AG, Goodman, LS, Rall, TW, Murad, F, Eds.; MacMillan: New York, 1985; Volume 21, pp. 926–957. [Google Scholar]
- Gröger, GD; Floss, HG. Biochemistry of ergot alkaloids – Achievements and challenges. Alkaloids 1998, 50, 171–218. [Google Scholar]
- Mantegani, S; Brambilla, E; Varasi, M. Ergoline derivatives: receptor affinity and selectivity. II Farmaco 1999, 54, 288–296. [Google Scholar]
- Pertz, H; Eich, E. Ergot alkaloids and their derivatives as ligands for serotoninergic, dopaminergic, and adrenergic receptors. In Ergot: The genus claviceps; Kren, V, Cvak, L, Eds.; Harwood Academic Publishers: Amsterdam, 1999; pp. 411–440. [Google Scholar]
- Piccoli, F; Ruggeri, RM. Dopaminergic agonists in the treatment of Parkinson’s disease: A review. J. Neural Transm 1995, 45, 187–195. [Google Scholar]
- Rabey, JM. Second generation of dopamine agonists: Pros and cons. J. Neural Transm 1995, 45, 213–224. [Google Scholar]
- Lee, EY; Chen, H; Shepherd, KR; Lamango, NS; Soliman, KFA; Charlton, CG. The inhibitory role of methylation on the binding characteristics of dopamine receptors and transporter. Neurosci. Res 2004, 48, 335–344. [Google Scholar]
- Meschler, JP; Howlett, AC. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology 2001, 40, 918–926. [Google Scholar]
- Millan, MJ; Maiofiss, L; Cussac, D. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharmacol. Exp. Ther 2002, 303, 791–804. [Google Scholar]
- Spivak, D; Shea, KJ. Molecular imprinting of carboxylic acids employing novel functional macroporous polymers. J. Org. Chem 1999, 64, 4627–4636. [Google Scholar]
- Yu, C; Mosbach, K. Molecular imprinting utilizing an amide functional group for hydrogen bonding leading to highly efficient polymers. J. Org. Chem 1997, 62, 4057–4064. [Google Scholar]
- Suedee, R; Seechamnanturakit, V; Canyuk, B; Ovatlarnporn, C; Martin, GP. Temperature sensitive dopamine-imprinted (N,N-methylene-bis-acrylamide cross-linked) polymer and its potential application to the selective extraction of adrenergic drugs from urine. J. Chromatogr. A 2006, 114, 239–249. [Google Scholar]
- Umpleby, RJ, 2nd; Baxter, SC; Chen, Y; Shah, RN; Shimizu, KD. Characterization of molecularly imprinted polymers with the Langmuir-Freundlich isotherm. Anal. Chem 2001, 73, 4584–4591. [Google Scholar]
- Umpleby, RJ, 2nd; Baxter, SC; Rampey, AM; Rushton, GT; Chen, Y; Shimizu, KD. Characterization of of the heterogeneous binding site affinity distributions in molecularly imprinted polymers. J. Chromatogr. B 2004, 804, 141–149. [Google Scholar]
- Tureil, E; Perez-Conde, C; Martin-Esteban, A. Assessment of the cross-reactivity and binding sites characterisation of a propazine-imprinted polymer using the Langmuir-Freundlich isotherm. Analyst 2003, 128, 137–141. [Google Scholar]
- Pregenzer, JF; Alberts, GL; Bock, JH; Slightom, JL; Im, WB. Characterization of ligand binding properties of the 5-HT1D receptors cloned from chimpanzee, gorilla and rhesus monkey in comparison with those from the human and guinea pig receptors. Neurosci. Lett 1997, 235, 117–120. [Google Scholar]
- Perachon, S; Schwartz, J-C; Sokoloff, P. Functional potencies of new antiparkinsonian drugs at recombinant human dopamine D1, D2 and D3 receptor. Eur J. Pharmacol 1999, 366, 293–300. [Google Scholar]
- Turkewitsch, P; Wandelt, B; Darling, GD; Powell, WS. Fluorescent functional recognition sites through molecular imprinting. A polymer-based fluorescent chemosensor for aqueous cAMP. Anal. Chem 1998, 70, 2025–2030. [Google Scholar]
- Motulsky, H; Christopoulus, A. Fitting dose response curves, GraphPad Software Inc: USA, 2002.
- Cheng, Y. -C.; Prusoff, W.H. Relationship between inhibition constant (Ki) and concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [Google Scholar]
- Holman, RB; Angwin, P; Barchas, JD. Simultaneous determination of indole- and catecholamines in small brain regions in the rat using a weak cation exchange resin. Neuroscience 1976, 1, 147–150. [Google Scholar]
- Bradford, MA. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar]
- Warsh, JJ; Chiu, A; Godse, DD; Coscina, DV. Determination of picogram levels of brain serotonin by a simplified liquid chromatographic electrochemical detection assay. Br. Res. Bull 1979, 4, 567–570. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Particle size a) (μm) | Pore diameter (nm) | Pore volume b) (mL/g) | BET Surface area b) (m2/g) |
|---|---|---|---|---|
| NIP | 29.10 | 52.21 | 0.25 (0.009) | 201.9 (24.6) |
| DS-MIP | 22.23 | 18.36 | 0.21 (0.006) | 133.3 (17.5) |
| D-MIP c) | 19.93 | 30.17 | 0.24 (0.014) | 130.4 (34.4) |
| S-MIP | 19.43 | 21.09 | 0.31 (0.005) | 158.4 (14.8) |
| Polymer | Ligand | Bmax (μmol/g) | Kd1 (mM) | Kd2 (mM) |
|---|---|---|---|---|
| NIP | D | 0.52 ± 0.17 | 1.21 ± 0.23 | 0.55 ± 0.23 |
| S | 1.45 ± 0.60 | 3.33 ± 0.73 | 0.24 ± 0.09 | |
| DS-MIP | D | 3.10 ± 0.52 | 0.073 ± 0.002 | 10.41 ± 0.21 |
| S | 5.71 ± 0.32 | 0.027 ± 0.0003 | 1.67 ± 0.13 | |
| D-MIP | D | 3.48 ± 0.70 | 0.094 ± 0.028 | 1.25 ± 0.60 |
| S | 4.63 ± 1.32 | 0.196 ± 0.054 | 3.53 ± 1.02 | |
| S-MIP | D | 3.34 ± 0.74 | 0.278 ± 0.092 | 10.01 ± 0.36 |
| S | 4.65 ± 0.32 | 0.074 ± 0.012 | 14.49 ± 3.25 |
| DS-MIP | D-MIP | S-MIP | |
|---|---|---|---|
| D | |||
| Nt | 3.64 ± 0.01 μmol/g | 2.76 ± 0.19 μmol/g | 10.27 ± 0.66 μmol/g |
| a | 3.30 ± 0.01 mM−1 | 3.44 ± 0.05 mM−1 | 1.02 ± 0.12 mM−1 |
| Ko | 4.17 ± 0.01 mM−1 | 4.48 ± 0.04 mM−1 | 1.03 ± 0.16 mM−1 |
| m | 0.84 ± 0.01 | 0.83 ± 0.03 | 0.75 ± 0.02 |
| Limits of affinity distribution* | 0.09–9.5 mM−1 | 0.09–9.5 mM−1 | 0.19–9.5 mM−1 |
| R2 | 0.998 | 0.966 | 0.975 |
| S | |||
| Nt | 5.86 ± 0.01 μmol/g | 46.54 ± 16.81 μmol/g | 5.00 ± 0.37 μmol/g |
| a | 4.73 ± 0.08 mM−1 | 0.07 ± 0.01 mM−1 | 4.02 ± 0.16 mM−1 |
| Ko | 12.85 ± 1.09 mM−1 | 0.03 ± 0.001 mM−1 | 8.08 ± 0.36 mM−1 |
| m | 0.61 ± 0.02 | 0.82 ± 0.02 | 0.67 ± 0.01 |
| Limits of affinity distribution* | 0.21–25.0 mM−1 | 0.19–9.5 mM−1 | 0.20–14.18 mM−1 |
| R2 | 0.976 | 0.993 | 0.997 |
| Ergot | Ki (μM)
| |||
|---|---|---|---|---|
| D ligand | S ligand | |||
| DS-MIP a) | Rat hypothalamus b) | DS-MIP a) | Rat hypothalamus b) | |
| Terguride | 0.14 ± 0.01 | 0.00003 ± 0.000005 | 7.79 ± 0.17 | 0.0013 ± 0.0001 |
| Pergolide | 0.20 ± 0.01 | 0.0007 ± 0.0001 | 12.78 ± 0.09 | 0.0060 ± 0.0002 |
| Agroclavine | 0.40 ± 0.01 | 0.0015 ± 0.0002 | 5.31 ± 0.22 | 0.0012 ± 0.0001 |
| Ergonovine | 69.57 ± 0.51 | 0.0025 ± 0.0001 | 14.74 ± 0.29 | 0.0012 ± 0.0001 |
| Ergocryptine | 124.88 ± 1.22 | 0.0028 ± 0.0002 | 24.85 ± 0.69 | 0.0100 ± 0.0004 |
| Ergocristine | 141.08 ± 1.47 | 0.0076 ± 0.0001 | 19.33 ± 0.60 | 0.0068 ± 0.0002 |
| Ergocornine | 216.60 ± 2.59 | 0.0104 ± 0.0002 | 27.25 ± 0.78 | 0.0200 ± 0.0001 |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/). This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suedee, R.; Seechamnanturakit, V.; Suksuwan, A.; Canyuk, B. Recognition Properties and Competitive Assays of a Dual Dopamine/Serotonin Selective Molecularly Imprinted Polymer. Int. J. Mol. Sci. 2008, 9, 2333-2356. https://doi.org/10.3390/ijms9122333
Suedee R, Seechamnanturakit V, Suksuwan A, Canyuk B. Recognition Properties and Competitive Assays of a Dual Dopamine/Serotonin Selective Molecularly Imprinted Polymer. International Journal of Molecular Sciences. 2008; 9(12):2333-2356. https://doi.org/10.3390/ijms9122333
Chicago/Turabian StyleSuedee, Roongnapa, Vatcharee Seechamnanturakit, Acharee Suksuwan, and Bhutorn Canyuk. 2008. "Recognition Properties and Competitive Assays of a Dual Dopamine/Serotonin Selective Molecularly Imprinted Polymer" International Journal of Molecular Sciences 9, no. 12: 2333-2356. https://doi.org/10.3390/ijms9122333