Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors

Department of Physics, Faculty of Sciences and Arts, Harran University, Osmanbey Campus, Sanliurfa 63300, Turkey
Int. J. Mol. Sci. 2008, 9(2), 181-197; https://doi.org/10.3390/ijms9020181
Submission received: 7 January 2008
/
Accepted: 17 January 2008
/
Published: 26 February 2008
(This article belongs to the Section Physical Chemistry, Theoretical and Computational Chemistry)
Abstract
:In the present study, quantitative structure–activity-relationship (QSAR) study on a group of sulfonamide Schiff-base inhibitors of Carbonic Anhydrase (CA) enzyme has been carried out using Codessa Pro methodology and software. Linear regression QSAR models of the biological activity (Ki) of 38 inhibitors of carbonic anhydrase CA-II isozyme were established with 12 different molecular descriptors which were selected from more than hundreds of geometrical, topological, quantum-mechanical, and electronic types of descriptors and calculated using Codessa Pro software. Among the models presented in this study, statistically the most significant one is a five-parameter equation with correlation coefficient, R2 values of ca. 0.840, and the cross-validated correlation coefficient, R2 values of ca. 0.777. The obtained models allowed us to reveal some physicochemical and structural factors, which are strongly correlated with the biological activity of the compounds.
1. Introduction
The metallo-protein carbonic anhydrase (CA, EC 4.2.1.1) is one of the most widely spread biological catalysts all over the phylogenetic tree. In humans, isozymes I, II, and IV are involved in respiration and regulation of the acid/base homeostasis. These complex processes involve both the transport of CO2/bicarbonate between metabolizing tissues and excretion sites (lungs, kidneys), facilitate CO2 elimination in capillaries and pulmonary microvasculature, eliminate H+ ions in the renal tubules and collecting ducts, as well as help in the reabsorption of bicarbonate in the brush border and thick ascending Henle loop of the kidneys. By producing the bicarbonate-rich aqueous humor secretion (mediated by ciliary processes isozymes CA I, II, CA IV and CA XII) within the eye, CAs are involved in vision, and their misfunctioning leads to high intraocular pressure and glaucoma. CA II is also involved in the bone development and in functions such as the differentiation of osteoclasts or the provision of acid for bone resorption in osteoclasts [1–4]. The presence of these isozymes in so many tissues and in a number of different isoforms represents an attractive objective for the design of inhibitors with biomedical applications.
Quantitative structure activity relationship (QSAR) studies are tools for predicting endpoints of interest in organic molecules acting as drugs [5]. Many physiological activities of a molecule can be related to their composition and structure. Molecular descriptors, which are numerical representations of the molecular structures, are used for performing QSAR analysis [6]. Sulfonamides represent the most important class of biologically active compounds as inhibitors of CAs. In the literature, there have been a number of QSAR studies of sulfonamides using quantum chemical [7–14] and topological [15–24] descriptors and 3-D approach of CoMFA and CoMSIA [25]. To the best of our knowledge, four QSAR studies [26–28, 10] have been carried out using sulfonamides with Schiff base as Carbonic anhydrase inhibitors.
In this study, we investigated QSAR for 38 sulfanilamide Schiff's base inhibitors of the physiologically relevant isozyme CAII using Codessa Pro approach [29]. To the best of our knowledge, all QSAR studies using sulfonamides with Schiff base have been performed on the same data set. The majority of molecules in our set have been newly synthesized and till now they have not been conducted in any QSAR study. The results of this study may help estimate the inhibition activity of sulfonamide with Schiff base of this series, prior to synthesis.
2. Results and Discussion
2.1. Computational details
For all the molecules studied, 3-D modeling and calculations of quantum mechanical descriptors were performed using the Gaussian 03 quantum chemistry package [30]. To save computational time, initial geometry optimizations were carried out with the molecular mechanics (MM) method, using the MM+ force fields. The lowest energy conformations of the molecules obtained by the MM method were further optimized by the DFT [31] method by employing Becke's three-parameter hybrid functional (B3LYP) [32] and the 6–31G (d) basis set. Their fundamental vibrations were also calculated using the same method to check if there were true minima. All the computations were carried out for the ground states of these molecules as single states. Codessa Pro was used for statistical analysis. This code uses diverse statistical structure property/activity correlation techniques for the analysis of experimental data in combination with the calculated molecular descriptors. It is worthy to mention here that we used a high level of theory (DFT/B3LYP) to obtain more precise data of descriptors during the calculation of the optimized 3-D geometry and quantum mechanical descriptors of the compounds, although no geometric, quantum mechanical, and thermo dynamical descriptors were involved in the obtained models presented in the following section. The heuristic method (HM) [33] implemented in Codessa Pro was employed for selecting the ‘best’ regression model. HM can either quickly give a good estimation about what quality of correlation to expect from the data, or can derive several best regression models. Besides, it will demonstrate as to which descriptors have bad or missing values, which descriptors are insignificant, and which descriptors are highly inter-correlated. This information will be helpful in reducing the number of descriptors involved in the search for the best QSAR model. A pre-selection of descriptors is accomplished by HM as follows. All descriptors are checked to ensure that (a) value of each descriptor is available for every structure and (b) there is a variation in these values. The descriptors for which values are not available for every structure in the data in question are discarded. Descriptors having a constant value for all structures in the data set are also discarded. A printout showing descriptors thus discarded is provided. Thereafter, the one-parameter correlation equations for each descriptor are calculated. To further reduce the number in the ‘‘starting set’’ of descriptors, the following criteria are applied and a descriptor is eliminated if: (a) the F-test's value for the one-parameter correlation with the descriptor is below 1.0, (b) the squared correlation coefficient of the one-parameter equation is less than R2 min 0.01 by default, (c) the parameter's t-value is less than t1 (where R2 min 0.1 by default and t1 1.5 by default are user-defined values), and (d) the descriptor is highly inter-correlated (above rfull, where rfull is a user-specified value by default 0.80), with another descriptor. All the remaining descriptors are then listed in the decreasing order according to the correlation coefficient of the corresponding one-parameter correlation equation. All two parameter regression models with remaining descriptors are developed and ranked by the regression correlation coefficient R2. A stepwise addition of further descriptor scales is performed to find the best multi-parameter regression models with optimum values of statistical criteria (highest values of R2, the cross-validated, R2 CV, and the F value). In addition to the descriptors calculated using Gaussian 03 and Codessa Pro, we have also added two physicochemical properties of compounds, namely, logarithm of 1-octonal/water partition coefficient (logP) and logarithm of aqueous solubility (logS) to the descriptor pool. These two parameters were calculated using WEB tool of ALOGPS 2.1 software [34]. This WEB tool calculates logP and logS using six different softwares and algorithms including ALOGPS 2.1. The calculation results of the properties are listed in the WEB for each software and in the average of six softwares as well. We have used average values of logP and logS from six different softwares in the regression procedures.
2.2. Results
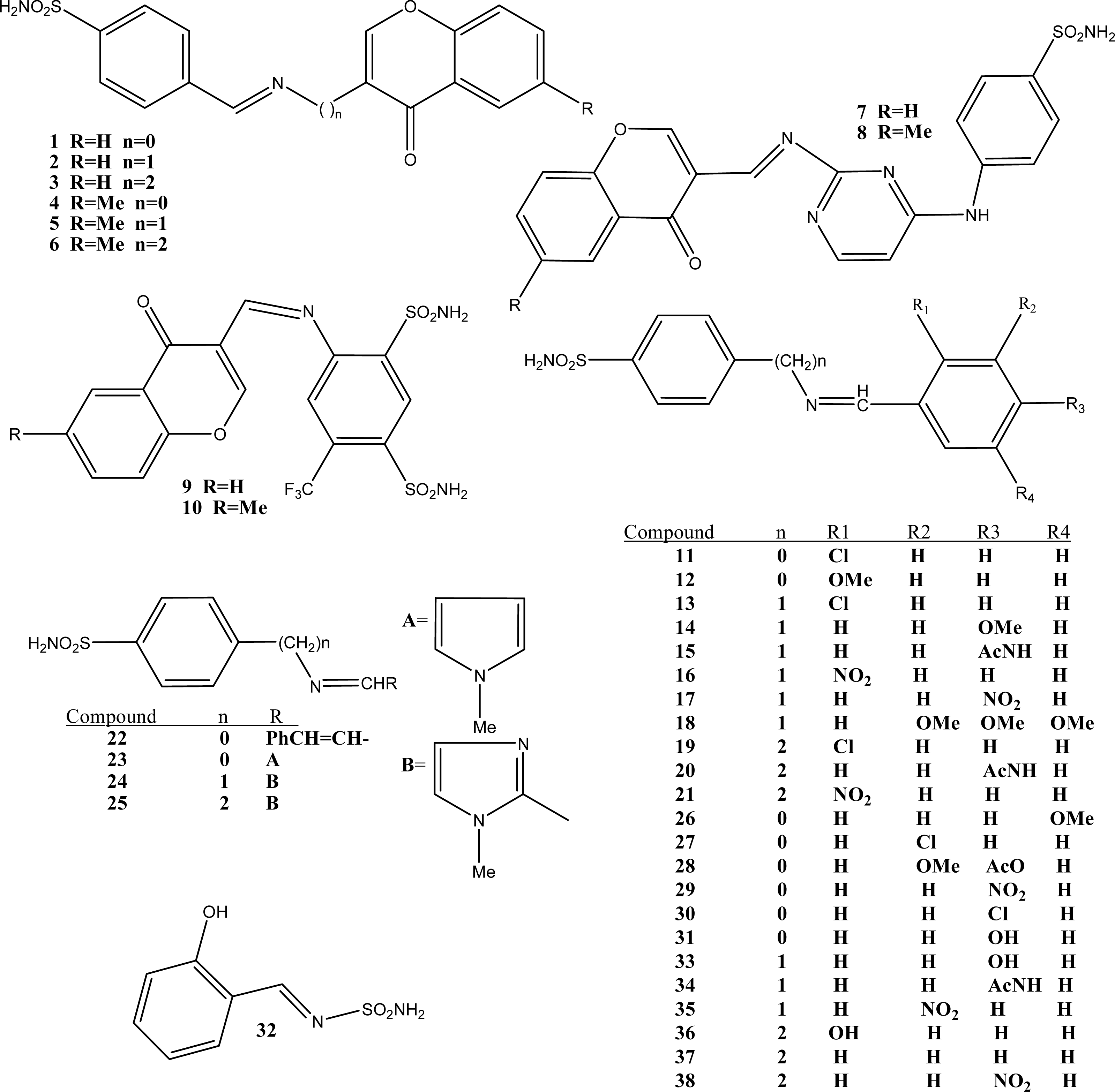
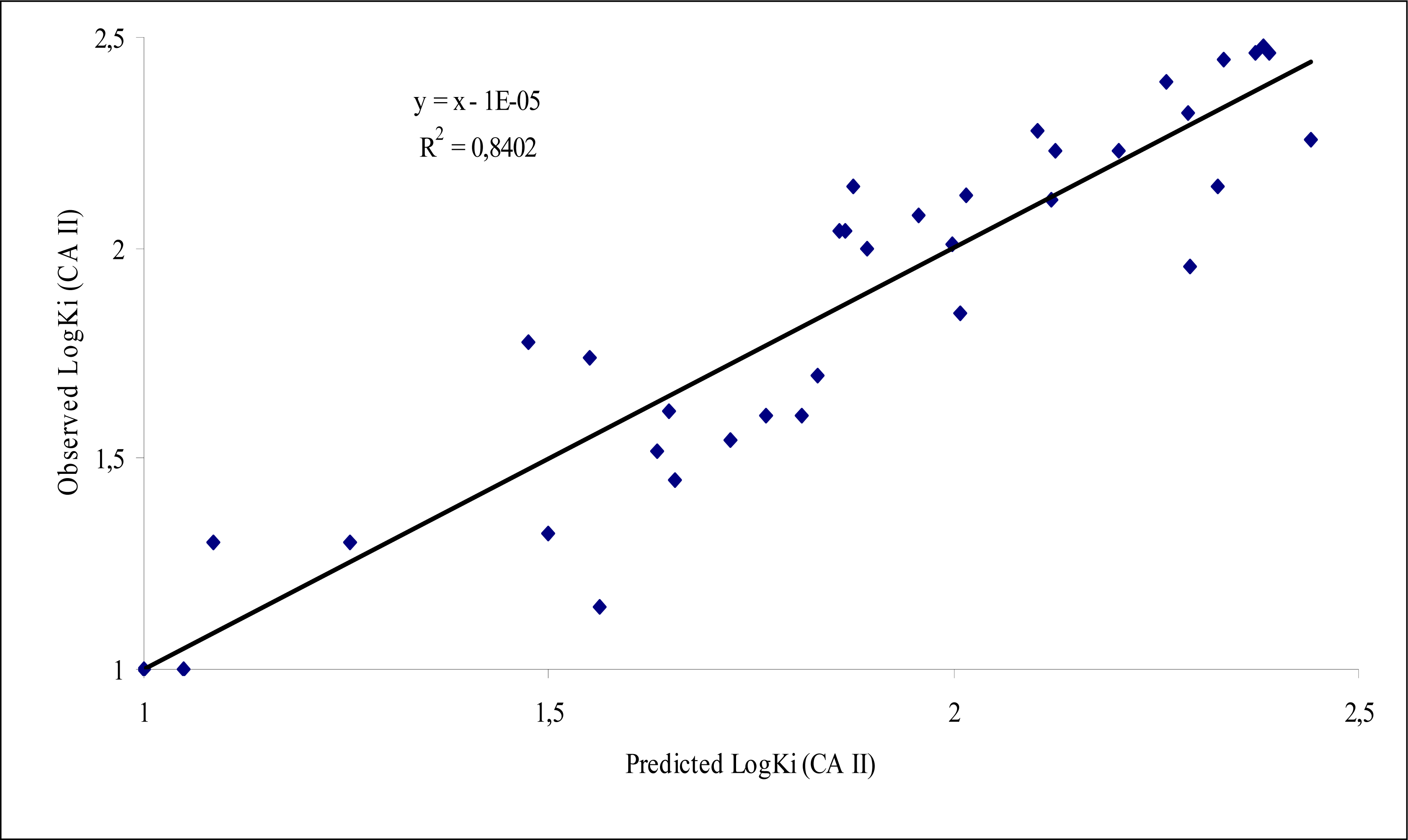
The structures of 38 Schiff-base sulfonamide compounds are shown in Figure 1. The experimental inhibitory activity of the compounds against CA II isozyme was taken from three references [35–37]. Table 1 shows the following information: (i) calculated molecular descriptor values involved in the models, (ii) experimental Ki (nM) values taken from the original references, and (iii) the calculated Ki (nM) values using the best model 5 obtained in this study. The plot of observed versus calculated Ki for CA II using model 5 is shown in Figure 2. The inter-correlation of descriptors is shown in Table 3.
Using the HM, several regression equations were obtained in this study. Among the regression results, five equations (the best one, two, three, four, and five parameters) were selected as models and are given in Table 2. In these models, the correlation coefficient, R2, is a measure of the fit of the regression equation. F, the Fisher test value, reflects the ratio of the variance explained by the model and the variance due to the error in the model. Higher values of F-test indicate the significance of the equation. s2 is the standard deviation of the regression. R2CV, the ‘leave one out’ (LOO) cross-validated coefficient, is a practical and reliable method for testing the predictive performance and stability of a regression model. LOO approach involves developing a number of models, with one sample omitted at a time. After developing each model, the omitted data are predicted and the differences between the experimental and predicted activity values are calculated. The R2CV values are then calculated according to the following formula [38]:
where yi is the actual experimental activity, y the average actual experimental activity, and ŷ î the predicted activity of compound i computed by the new regression equation obtained each time after leaving out one datum point (No. i).
In the present work, more than two hundred descriptors were exploited. In Codessa Pro, descriptors are divided into groups such as constitutional, topological, geometrical, electrostatic, quantum chemical, thermodynamic, and constructed. Constitutional descriptors are related to the number of atoms and bonds in each molecule. Topological descriptors include valence and non valence molecular connectivity indices calculated from the hydrogen-suppressed formula of the molecule, encoding information about the size, composition, and the degree of branching of a molecule. Geometrical descriptors are calculated from 3-D atomic coordinates of the molecule and comprise moments of inertia, shadow indices, molecular volumes, molecular surface areas, and gravitation indices. Electrostatic descriptors reflect characteristics of the charge distribution of the molecule. Quantum chemical descriptors encode the polar interactions between molecules or their chemical reactivity and the activation energy of the corresponding chemical reaction. Thermodynamic descriptors are quantum mechanically calculated on the basis of the total partition function of the molecule Q and its electronic, translational, rotational, and vibrational components. Codessa Pro also allows one to construct new descriptors by using the existing descriptors. In this way, the author has constructed some common quantum chemical indices, namely, chemical hardness, electronegativity, and electrophilicity from HOMO and LUMO orbital energies. The results shown in Table 2 have been quite surprising, which is attributed to the fact that no quantum chemical indices has turned out in our models. In our previous studies [13–14], the QSAR models have been drawn up from the quantum mechanical descriptors of a group of diverse aromatic and heterocyclic sulfonamides and from the inhibitory activity of these compounds against CA II isozyme. For comparison, we have tried to correlate inhibitory activity Ki-CA II of molecule set of this study (Schiff base sulfonamides) with the same quantum mechanical descriptors involved in QSAR models in our previous works. The correlation coefficient was very poor, less than (R< 0.1). This result indicates that inhibition mechanism of Schiff-base sulfonamides is different from that of the aromatic and heterocyclic sulfonamides.
According to the preliminary regression analysis, these two compounds exhibited unusual behaviors in all the models. When the heuristic method has been run with default for 38 compounds, the best one, two, three, four and five parameter equations have shown up as the program output. In all these five equations, compounds 29 and 38 have had the largest standard residual (almost twice of mean residua). After selecting these two compounds as outliers, the statistical quality of one, two, three, four and five parameter equations were increased dramatically such as statistical parameters for five parameter equation R2 from 0.71 to 0.84, F from 15.96 to 31.54, and s2 from 0.061 to 0.034. It is worthy here mention that the descriptors involved in the best equations obtained for 38 compounds set and 36 compounds set are not the same. The best one, two, three, four and five parameter equations obtained from 36 compounds are presented as models in following.
A perusal of Table 2 shows that twelve types of descriptors are involved in all the five models. The use of HM method yielded the best one-parameter regression expression as follows.
Here and thereafter, N is the number of compounds, R2 is the correlation coefficient, R2 CV is the ‘leave one out’ (LOO) cross-validated coefficient, F is the Fisher-statistic value, and s2 is the standard deviation of the regression equation. In this model, as well as the models presented below, compounds 29 and 38 are outliers. In this model, RNBR is the relative number of benzene rings. RNBR is the ratio between the numbers of benzene rings divided by the total number of atoms in the molecules. In the above equation, RNBR has a positive-sign coefficient. This means that increases in the magnitude of RNBR favors the exhibitions of the inhibitory activity of CA II-Ki.
Among the obtained two-parameter models, statistically the best one is as below:
In this model, NBR is the number of benzene rings and NCA is the number of C atoms in the molecules. These two descriptors are constitutional. NBR has a coefficient with positive sign and NCA has a coefficient with a negative sign. When models 1 and 2 are considered together, both models highlight the same features of the molecules. According to the models, substituting benzene with nitro or hydroxy groups instead of methyl groups and decreasing the number of CH2 bond to imine nitrogen are favorable for an increase in the inhibition activity of the compounds.
Among the obtained three-parameter models, statistically the best one is as follows:
In this model, RNDB is the relative number of double bonds, DPSA-1 is defined as the difference in CPSAs (PPSA1 (partial positive surface area) -PNSA1 (partial negative surface area)) [Zefirov's PC] and MiPCN is the Min partial charge for a N atom [Zefirov's PC]. RNDB is a constitutional descriptor and has a coefficient with a negative sign. A decrease in the magnitude of RNDB favors the exhibitions of the inhibitory activity of CA II-Ki. DPSA-1 is an electrostatically charged partial surface-area descriptor and encodes features responsible for polar interactions between molecules. The negative sign of coefficient of DPSA-1 highlights a decrease in the magnitude of RNDB, which favors the exhibitions of the inhibitory activity of CA II-Ki. MiPCN is an electrostatic descriptor and reflects minimum partial charge on the N atom in the molecules. The positive sign of the coefficient of MiPCN indicates that increases in the magnitude of MiPCN are favorable for an increase of inhibition activity of the compounds.
Among the obtained four-parameter models, statistically the best one is as shown below:
In this model, ALS is the Average LogS, 3χ is the Randic index (order 3), RPCG is the Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC], and RNDB is the relative number of double bonds as in model 4. ALS is the logarithm of aqueous solubility of the compounds. The negative sign of the coefficient of ALS highlights a decrease in the magnitude of ALS, which favors the exhibitions of inhibitory activity of CA II-Ki. 3χ is a topological descriptor, and describes the atomic connectivity and branching information of the molecules. 3χ has a coefficient with a negative sign. This means that the third-order branching is not the favorable parameter for the exhibition of the inhibitory activity. RPCG, the RPCG Relative positive charge (QMPOS/QTPLUS) is an electrostatic descriptor, and reflects characteristics of the charge distribution of the molecules. The positive sign of the coefficient of RPCG indicates that increases in the magnitude of relative positive charge of molecules are favorable for an increase of the inhibition activity of the compounds.
During our regression analysis using HM, we obtained several five-parameter equations. Out of these five-parameter equations, equation 5 consisting of NBR the Number of benzene rings, NCA the Number of C atoms, NNA the Number of N atoms, ALP the Average logP, and 2AIC the Average information content (order 2) is the best. This model is as shown below:
In this model, the sign of coefficients of NBR and NCA is the same as in that in the model 2 and thus they carry the same significance as in that model. NNA is a constitutional descriptor. It has a positive sign of coefficient. This means that the increase in the magnitude of NNA is the favorable parameter for the exhibition of the inhibitory activity. ALP is the logarithm of 1-octonal/water partition coefficient and exhibits hydrophobicity of compounds. The positive sign of coefficient of ALP indicates that Log CA II-Ki increases with increase in the magnitude of Average logP. 2AIC is the second-order average complementary information content that could be considered an index of heterogeneity of a molecule [38]. The positive sign of coefficient of 2AIC indicates that increases in the magnitude of 2AIC are favorable for an increase of inhibition activity of the compounds.
By default setting, HM uses maximum five descriptors to construct a regression model. We have changed the default setting of the software in order to allow for the use of more than five descriptors. This has yielded several six- and seven-parameter regression equations. None of those equations has better statistic parameters than the above five parameters (equation 5). Although R2 values of those six- and seven-parameter equations were greater than those of equation 5, they were not selected as models in this study due to their relatively low F value.
2.3. Discussion
Twelve types of descriptors were involved in the models. Two of the descriptors are physicochemical property, namely, ALS, average logS, and ALP, average logP. In model 4, average logS negatively contribute to the inhibitory activity. In model 5, average logP positively contributes to the inhibitory activity. This result is expected due to the fact that the average logP is inversely proportional to the average logS. Five of the involved descriptors are constitutional. RNBR, the relative number of benzene rings in model 1, and NBR, the number of benzene rings in the models 2 and 5 positively contribute to the inhibitory activity. On the contrary, NCA, the number of C atoms, negatively contribute to the inhibitory activity in the models 2 and 3. When these two results are combined together, one could draw a conclusion that one should avoid substituting benzene rings with C-containing groups and adding CH2 bond to imine nitrogen for designing Schiff-base sulfonamide compounds with increased inhibitory activity. Another constitutional descriptor is NNA, the number of N atoms which positively contribute to the inhibitory activity in model 5. As a consequence, substituting benzene ring with nitro groups or inserting an N-containing ring to a Schiff-base sulfonamide compound may help increase the inhibitory activity. Two descriptors, 3χ and 2AIC, out of twelve are topological index. 3χ is the third-order Randic index which negatively contributes to the inhibitory activity in model 4. This means that if one wants to design a Schiff-base sulfonamide with high inhibitory activity, it should be considered that the third-order branching is not the favorable parameter. Another topological index is 2AIC, the second-order average information content. This descriptor reflects the heterogeneity of a molecule and positively contributes to the inhibitory activity in model 5. Remaining three descriptors are electrostatic index, and they characterize the charge distribution of the molecules. MiPCN, Min partial charge for a N atom [Zefirov's PC], and RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC], positively contribute to the inhibitory activity in models 3 and 4, respectively. DPSA-1, Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC], negatively contributes to the inhibitory activity in model 3. The values of these electrostatic indexes of molecules are affected by substituting groups. This should be taken into account when one tries to design a new molecule.
2.4. Conclusion
In the present study, the structural descriptors of 36 sulfonamide Schiff-base inhibitors of Carbonic Anhydrase CA II enzyme have been correlated with the their inhibition constant Ki using Codessa Pro methodology. Among the obtained model, five-parameter equation consists of the descriptors namely the Number of benzene rings, the Number of C atoms, the Number of N atoms, the Average logP, and the Average information content (order 2) is the best one. The statistical parameters of this model are the R2 = 0.84, R2 CV=0.77, F= 31.54, and s2 = 0.034.
3. Experimental Section
Inhibition of CA II
Topological indexes
Two topological indexes were involved in our models. These two indices are defined using the formula given below according to Codessa Pro reference Manual [33].
Randic and Kier & Hall indices (order 0–3). The general formula for the calculation of these indexes is as follows:
where δi and δj (i ≠ j) correspond to the coordination numbers of atoms (Randic index [40]) or to the values of the atomic connectivity (valence connectivity index nχυ by Kier and Hall [41]). The atomic connectivity for the i-th atom in the molecular skeleton is calculated using the formula:
where Zi is the total number of electrons in the i-th atom,
is the number of valence electrons, and Hi is the number of hydrogens directly attached to the i-th atom.
Information content index and its derivatives (order 0–2). The average information content is defined on the basis of the Shannon information theory and is calculated as follows [42–43]:
where ni is a number of atoms in the i-th class and n is a total number of atoms in the molecule. The division of atoms into different classes depends upon the coordination sphere taken into account. This leads to the indices of different order k. The information content (IC) is equal to average information content multiplied by the total number of atoms. Other information content indices (SIC – structural IC, CIC – complementary IC, and BIC – bonding IC) are defined as follows [44]:
where q is a number of edges in the structural graph of the molecule.
Electrostatic indexes
According to Codessa Pro reference Manual [33], electrostatic indexes were involved in our models and are calculated using the formulae given below.
Charged partial surface-area descriptors. Charged partial surface-area (CSPA) descriptors have been invented by Jurs et al. [45–46] in terms of the whole surface area of the molecule and in terms of functional group portions.
Descriptors involved in this study are as follows:
- DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC].
- RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC].
Minimum and maximum partial charges for particular types of atoms (e.g. C, O, N etc.). The empirical partial charges in the molecule are calculated using the approach proposed by Zefirov [47–48]. This method is based on the Sanderson electronegativity scale and uses the concept which represents the molecular electronegativity as a geometric mean of atomic electronegativities.
Acknowledgments
This work has been supported by Harran University Research Council (HUBAK) Project no: 788.
References and Notes
- Lehtonen, JM; Parkkila, S; Vullo, D; Casini, A; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozyme XIII with aromatic and heterocyclic sulfonamides: a novel target for the drug design. Bioorg Med Chem Lett 2004, 14, 3757–3762. [Google Scholar]
- Supuran, CT; Scozzafava, A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007, 15, 4336–4350. [Google Scholar]
- Carbonic Anhydrase Its Inhibitors and Activators; Supuran, CT; Scozzafava, A; Conway, J (Eds.) CRC Press: Boca Raton, FL, USA, 2004; pp. 1–363, and references cited therein.
- Parkkila, S. An Overview of the Distribution and Function of Carbonic Anhydrase in Mammals. In The Carbonic Anhydrases—New Horizons; Chegwidden, WR, Edwards, Y, Carter, N, Eds.; Birkhäuser Verlag: Basel, Switzerland, 2000; pp. 79–93. [Google Scholar]
- Karelson, M. Molecular Descriptors in QSAR/QSPR; Wiley: New York, NY, 2000. [Google Scholar]
- Tadeschini, R; Consonni, V. Handbook of Molecular Descriptors; Wiley–VCH: Weinheim, Germany, 2000. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 86. A QSAR study on some sulfonamide drugs which lower intra-ocular pressure, using the ACE non-linear statistical method. Eur J Med Chem 2000, 35, 859–865. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 61. Quantum chemical QSAR of a group of benzenedisulfonamides. Eur J Med Chem 1999, 34, 463–474. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors – Part 57: Quantum chemical QSAR of a group of 1,3,4-thiadiazole- and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem 1999, 34, 41–50. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors-Part 47: Quantum chemical quantitative structure-activity relationships for a group of sulfanilamide Schiff base inhibitors of carbonic Anhydrase. Eur J Med Chem 1998, 33, 489–500. [Google Scholar]
- Clare, BW; Supuran, CT. Carbonic anhydrase inhibitors. Part 41. Quantitative structure-activity correlations involving kinetic rate constants of 20 sulfonamide inhibitors from a non-congeneric series. Eur J Med Chem 1997, 32, 311–319. [Google Scholar]
- Supuran, CT; Clare, BW. Carbonic anhydrase inhibitors. Part 24. A quantitative structure-activity relationship study of positively charged sulfonamide inhibitors. Eur J Med Chem 1995, 30, 687–696. [Google Scholar]
- Eroglu, E; Türkmen, H. A DFT-based quantum theoretic QSAR study of aromatic and heterocyclic sulfonamides as carbonic anhydrase inhibitors against isozyme, CA-II. Journal of Molecular Graphics and Modelling 2007, 26, 701–708. [Google Scholar]
- Eroglu, E; Türkmen, H; Güler, S; Palaz, S; Oltulu, O. A DFT-Based QSARs study of Acetazolamide/Sulfanilamide derivatives with carbonic anhydrase (CA-II) isozyme inhibitory activity. Int J Mol Sci 2007, 8, 145–155. [Google Scholar]
- Agrawal, VK; Singh, J; Khadikar, PV; Supuran, CT. QSAR study on topically acting sulfonamides incorporating GABA moieties: A molecular connectivity approach. Bioorg Med Chem Lett 2006, 16, 2044–2051. [Google Scholar]
- Melagraki, G; Afantitis, A; Sarimveis, H; Iglessi-Markopoulou, O; Supuran, CT. QSAR study on para-substituted aromatic sulfonamides as carbonic anhydrase II inhibitors using topological information indices. Bioorg Med Chem 2006, 14, 1108–1114. [Google Scholar]
- Agrawal, VK; Banerji, M; Gupta, M; Singh, J; Khadikar, PV; Supuran, CT. QSAR study on carbonic anhydrase inhibitors: water-soluble sulfonamides incorporating β-alanyl moieties, possessing long lasting-intra ocular pressure lowering properties-a molecular connectivity approach. Eur J Med Chem 2005, 40, 1002–1012. [Google Scholar]
- Khadikar, PV; Sharma, V; Karmarkar, S; Supuran, CT. QSAR studies on benzene sulfonamide carbonic anhydrase inhibitors: need of hydrophobic parameter for topological modeling of binding constants of sulfonamides to human CA-II. Bioorg Med Chem Lett 2005, 15, 923–930. [Google Scholar]
- Jaiswal, M; Khadikar, PV; Supuran, CT. Topological modeling of lipophilicity, diuretic activity, and carbonic inhibition activity of benzene sulfonamides: a molecular connectivity approach. Bioorg Med Chem Lett 2004, 14, 5661–5666. [Google Scholar]
- Agrawal, VK; Bano, S; Supuran, CT; Khadikar, PV. QSAR study on carbonic anhydrase inhibitors: aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties, with topical activity as antiglaucoma agents. Eur J Med Chem 2004, 39, 593–600. [Google Scholar]
- Jaiswal, M; Khadikar, PV; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors: the first QSAR study on inhibition of tumor-associated isoenzyme IX with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2004, 14, 3283–3290. [Google Scholar]
- Thakur, A; Thakur, M; Khadikar, PV; Supuran, CT; Sudele, P. QSAR study on benzenesulphonamide carbonic anhydrase inhibitors: topological approach using Balaban index. Bioorg Med Chem 2004, 12, 789–793. [Google Scholar]
- Agrawal, VK; Sharma, R; Khadikar, PV. QSAR Studies on Carbonic Anhydrase Inhibitors: A Case of Ureido and Thioureido Derivatives of Aromatic/Heterocyclic Sulfonamides. Bioorg Med Chem 2002, 10, 2993–2999. [Google Scholar]
- Khadikar, PV; Clare, BW; Balaban, AT; Supuran, CT; Agrawal, VK; Singh, J; Joshi, AK; Lakwani, M. QSAR modeling of carbonic anhydrase-I, -II and -IV inhibitory activities: Relative correlation potential of six topological indices. Revue Roumaine De Chimie 2006, 51, 703–717. [Google Scholar]
- Huang, H; Pan, X; Tan, N; Zeng, N; Ji, C. 3D-QSAR study of sulfonamide inhibitors of humancarbonic anhydrase II. Eur J Med Chem 2007, 42, 365–372. [Google Scholar]
- Agrawal, VK; Singh, J; Pandey, A; Khadikar, PV. Modelling of inhibitory activities of sulphanilamide Schiff bases using physicochemical properties. Oxidation Communications 2006, 29, 803–816. [Google Scholar]
- Supuran, CT; Clare, BW. Orbital symmetry in QSAR: Some Schiff's base inhibitors of carbonic anhydrase. SAR and QSAR in Environmental Research 2001, 12, 17–29. [Google Scholar]
- Agrawal, VK; Srivastava, S; Khadikar, PV; Supuran, CT. Quantitative Structure–Activity Relationship Study on Sulfanilamide Schiff's Bases: Carbonic Anhydrase (CA) Inhibitors. Bioorg Med Chem 2003, 11, 5353–5362. [Google Scholar]
- CODESSA (Comprehensive Descriptors for Structural and Statistical Analysis), Semichem, 7204, Mullen, Shawnee, KS 66216 USA, Copyright© Semichem and the University of Florida, 1995–2004.
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA, Jr; Vreven, T; Kudin, KN; Burant, JC; Millam, JM; Iyengar, SS; Tomasi, J; Barone, V; Mennucci, B; Cossi, M; Scalmani, G; Rega, N; Petersson, GA; Nakatsuji, H; Hada, M; Ehara, M; Toyota, K; Fukuda, R; Hasegawa, J; Ishida, M; Nakajima, T; Honda, Y; Kitao, O; Nakai, H; Klene, M; Li, X; Knox, JE; Hratchian, HP; Cross, JB; Adamo, C; Jaramillo, J; Gomperts, R; Stratmann, RE; Yazyev, O; Austin, AJ; Cammi, R; Pomelli, C; Ochterski, JW; Ayala, PY; Morokuma, K; Voth, GA; Salvador, P; Dannenberg, JJ; Zakrzewski, VG; Dapprich, S; Daniels, AD; Strain, MC; Farkas, O; Malick, DK; Rabuck, AD; Raghavachari, K; Foresman, JB; Ortiz, JV; Cui, Q; Baboul, AG; Clifford, S; Cioslowski, J; Stefanov, BB; Liu, G; Liashenko, A; Piskorz, P; Komaromi, I; Martin, RL; Fox, DJ; Keith, T; Al-Laham, MA; Peng, CY; Nanayakkara, A; Challacombe, M; Gill, PMW; Johnson, B; Chen, W; Wong, MW; Gonzalez, C; Pople, JA. Gaussian 03, Revision B05; Gaussian, Inc: Pittsburgh, PA, 2003. [Google Scholar]
- Parr, RG; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, 1989. [Google Scholar]
- Becke, AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993, 98, 5648–5648. [Google Scholar]
- CODESSATM, References Manual, V. 2.13 (PC)., Semichem, 7204, Mullen, Shawnee, KS, USA, Copyright Semichem and the University of Florida, 2002.
- Drug Discov Today. 2005, 10, pp. 1497–500. (a) http://www.vcclab.org/lab/alogPs/start.html. (b) Tetko, I. V. Computing chemistry on the web.
- Supuran, CT; Scozzafava, A; Popescu, A; Bobes-Tureac, R; Banciu, A; Bobes-Tureac, G; Bamciu, MD. Carbonic anhydrase inhibitors. Part 43. Schiff bases derived from aromatic sulfonamides: towards more specific inhibitors for membrane-bound versus cytosolic isozymes. Eur J Med Chem 1997, 32, 445–452. [Google Scholar]
- Krungkrai, J; Scozzafava, A; Reungprapavut, R; Krungkrai, SR; Rattanajak, R; Kamchonwongpaisand, S; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: towards antimalarials with a novel mechanism of action. Bioorg Med Chem 2005, 13, 483–489. [Google Scholar]
- Puccetti, L; Fasolis, G; Vullo, D; Chohan, ZH; Scozzafava, A; Supuran, CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, IX, and XII with Schiff's bases incorporating chromone and aromatic sulfonamide moieties, and their zinc complexes. Bioorg Med Chem Lett 2005, 15, 3096–3101. [Google Scholar]
- Mayers, RH. Classical and Modern Regression with Applications; PWSKENT Publ. Co: Boston, MA, 1990. [Google Scholar]
- Computational Chemical Graph Theory; Basak, S; Niemi, GJ; Veith, GD; Rouvray, DH (Eds.) Nova Science Publishers: New York, NY, 1990.
- Randic, M. Characterization of molecular branching. J Am Chem Soc 1975, 97, 6609–6615. [Google Scholar]
- Kier, LB; Hall, LH. Molecular Connectivity in Chemistry and Drug Reasearch; Academic Press: New York, NY, 1976. [Google Scholar]
- Kier, L. B. Use Of Molecular Negentropy To Encode Structure Governing Biological-Activity. J Pharm Sci 1980, 69, 807–810. [Google Scholar]
- Bonchev, D. Information Theoretic Indices for Characterization of Chemical Structure; Wiley-Interscience: New York, NY, 1983. [Google Scholar]
- Basak, SC; Harriss, DK; Magnuson, VR. Comparative-Study Of Lipophilicity Versus Topological Molecular Descriptors In Biological Correlations. J Pharm Sci 1984, 73, 429–437. [Google Scholar]
- Stanton, DT; Jurs, PC. Development And Use Of Charged Partial Surface-Area Structural Descriptors In Computer-Assisted Quantitative Structure Property Relationship Studies. Anal Chem 1990, 62, 2323–2329. [Google Scholar]
- Stanton, DT; Egolf, LM; Jurs, PC; Hicks, MG. Computer-Assisted Prediction Of Normal Boiling Points Of Pyrans and Pyrroles. J Chem Inf Comput Sci 1992, 32, 306–316. [Google Scholar]
- Zefirov, NS; Kirpichenok, MA; Ismailov, FF; Trofimov, MI. Calculation schemes for atomic electronegativities in molecular graphs within the framework of sanderson principle. Dokl Akad Nauk SSSR 1987, 296, 883–887. [Google Scholar]
- Kirpichenok, MA; Zefirov, NS. Electronegativity and geometry of molecules. 1. Principles of developed approach and analysis of the effect of nearest electrostatic interactions on the bond length in organic molecules. Zh Org Khim 1987, 23, 673–691. [Google Scholar]
Figure 1.
Molecular structure of Schiff-base sulfonamides used in the present study

Figure 2.
Description Plot of observed versus predicted LogKi CA II values using model 5.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated descriptors involved in the models and LogCA II-Ki of 38 sulfonamides compounds predicted by model 5.
| Comp. | RNBR | NBR | NCA | RNDB | DPSA-1 | MiPCN | ALS | 3χ | RPCG | NNA | ALP | 2AIC | Obs. Ki | Pre. Ki | Residue |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.0571 | 2.0000 | 16.0000 | 0.1351 | 21.4428 | -0.0912 | -4.0600 | 8.3122 | 0.0934 | 2.0000 | 1.4900 | 4.1579 | 1.447a | 1.6557 | 0.2087 |
| 2 | 0.0526 | 2.0000 | 17.0000 | 0.1250 | 68.0099 | -0.0967 | -4.1400 | 8.5538 | 0.0894 | 2.0000 | 1.6600 | 4.3006 | 1.146a | 1.5640 | 0.418 |
| 3 | 0.0488 | 2.0000 | 18.0000 | 0.1163 | 105.7782 | -0.0980 | -4.1300 | 8.8038 | 0.0855 | 2.0000 | 1.9700 | 4.4307 | 1.322a | 1.4996 | 0.1776 |
| 4 | 0.0526 | 2.0000 | 17.0000 | 0.1250 | 54.8184 | -0.0912 | -4.3100 | 8.6547 | 0.0902 | 2.0000 | 1.9000 | 4.3951 | 1.544a | 1.7243 | 0.1803 |
| 5 | 0.0488 | 2.0000 | 18.0000 | 0.1163 | 113.8093 | -0.0967 | -4.4600 | 8.8962 | 0.0865 | 2.0000 | 2.0800 | 4.4512 | 1.740a | 1.5507 | -0.1893 |
| 6 | 0.0455 | 2.0000 | 19.0000 | 0.1087 | 165.0231 | -0.0980 | -4.3200 | 9.1462 | 0.0828 | 5.0000 | 2.3900 | 4.5694 | 1.778a | 1.4747 | -0.3033 |
| 7 | 0.0444 | 2.0000 | 20.0000 | 0.1042 | 59.8359 | -0.1043 | -4.7700 | 10.8703 | 0.0693 | 5.0000 | 1.7700 | 4.6356 | 1.518a | 1.6333 | 0.1153 |
| 8 | 0.0417 | 2.0000 | 21.0000 | 0.0980 | 117.0900 | -0.1043 | -4.9300 | 11.2127 | 0.0675 | 3.0000 | 2.1800 | 4.8192 | 1.612a | 1.6494 | 0.0374 |
| 9 | 0.0465 | 2.0000 | 17.0000 | 0.1556 | -111.7607 | -0.0872 | -5.4600 | 10.5070 | 0.1504 | 3.0000 | 2.0600 | 4.4434 | 2.008a | 1.9995 | -0.0085 |
| 10 | 0.0435 | 2.0000 | 18.0000 | 0.1458 | -67.6019 | -0.0872 | -5.9000 | 10.8494 | 0.1474 | 2.0000 | 2.4600 | 4.6297 | 2.123a | 2.0154 | -0.1076 |
| 11 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | 31.6593 | -0.0932 | -4.2600 | 6.3063 | 0.1267 | 2.0000 | 2.3900 | 3.7736 | 2.461b | 2.3724 | -0.0886 |
| 12 | 0.0588 | 2.0000 | 14.0000 | 0.0857 | 130.0701 | -0.0935 | -3.7900 | 6.6081 | 0.1082 | 2.0000 | 1.9000 | 3.9476 | 2.230b | 2.1247 | -0.1053 |
| 13 | 0.0606 | 2.0000 | 14.0000 | 0.0882 | 20.6325 | -0.0986 | -4.3900 | 6.5479 | 0.1193 | 2.0000 | 2.4700 | 3.9535 | 1.954b | 2.2916 | 0.3376 |
| 14 | 0.0541 | 2.0000 | 15.0000 | 0.0789 | 138.1794 | -0.1001 | -3.9200 | 6.8399 | 0.1040 | 3.0000 | 1.9300 | 4.0539 | 2.079b | 1.9578 | -0.1212 |
| 15 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 111.5054 | -0.1002 | -3.8000 | 7.0673 | 0.0950 | 3.0000 | 1.2600 | 4.2531 | 2.041b | 1.8666 | -0.1744 |
| 16 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -18.8479 | -0.0984 | -4.0900 | 7.0830 | 0.1145 | 3.0000 | 1.8300 | 4.0436 | 2.477b | 2.3815 | -0.0955 |
| 17 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -22.6421 | -0.1000 | -4.1100 | 7.1305 | 0.1169 | 2.0000 | 1.8300 | 3.9864 | 2.146b | 2.3257 | 0.1797 |
| 18 | 0.0444 | 2.0000 | 17.0000 | 0.0652 | 291.3417 | -0.0991 | -3.9900 | 8.2996 | 0.0693 | 2.0000 | 1.6400 | 3.9908 | 1.301b | 1.2556 | -0.0454 |
| 19 | 0.0556 | 2.0000 | 15.0000 | 0.0811 | 65.0019 | -0.0999 | -4.4000 | 6.7979 | 0.1125 | 3.0000 | 2.8900 | 4.1144 | 2.322b | 2.2884 | -0.0336 |
| 20 | 0.0465 | 2.0000 | 17.0000 | 0.0909 | 140.9603 | -0.1015 | -3.9200 | 7.3173 | 0.0906 | 3.0000 | 1.6000 | 4.3855 | 1.602b | 1.8128 | 0.2108 |
| 21 | 0.0500 | 2.0000 | 15.0000 | 0.0732 | 122.8380 | -0.1001 | -4.0600 | 7.3330 | 0.1367 | 3.0000 | 1.6100 | 4.2719 | 2.397b | 2.2632 | -0.1338 |
| 22 | 0.0588 | 2.0000 | 15.0000 | 0.1143 | 40.6266 | -0.0956 | -3.7700 | 6.2710 | 0.1287 | 2.0000 | 2.5200 | 3.7556 | 1.698b | 1.8331 | 0.1351 |
| 23 | 0.0323 | 1.0000 | 12.0000 | 0.1250 | 90.8476 | -0.1147 | -2.5200 | 6.0563 | 0.1241 | 3.0000 | 0.6900 | 4.1682 | 1.000b | 1.0004 | 0.0004 |
| 24 | 0.0303 | 1.0000 | 12.0000 | 0.1176 | 155.8272 | -0.1064 | -2.1200 | 6.2979 | 0.1070 | 4.0000 | -0.3000 | 4.3549 | 1.301b | 1.0857 | -0.2153 |
| 25 | 0.0278 | 1.0000 | 13.0000 | 0.1081 | 209.1037 | -0.1064 | -2.3600 | 6.5479 | 0.1015 | 4.0000 | 0.1200 | 4.4823 | 1.000b | 1.0497 | 0.0497 |
| 26 | 0.0645 | 2.0000 | 13.0000 | 0.0938 | 43.8927 | -0.0934 | -3.2500 | 6.3063 | 0.1498 | 2.0000 | 1.5000 | 3.8574 | 2.230c | 2.2027 | -0.0273 |
| 27 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | -5.1570 | -0.0932 | -4.2600 | 6.3063 | 0.1267 | 2.0000 | 2.4500 | 3.7736 | 2.462c | 2.3894 | -0.0726 |
| 28 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 186.4766 | -0.0941 | -4.1900 | 7.6016 | 0.0993 | 2.0000 | 1.5200 | 4.3929 | 2.000c | 1.8939 | -0.1061 |
| 29d | 0.0625 | 2.0000 | 13.0000 | 0.0909 | -51.7829 | -0.0946 | -4.0500 | 6.8889 | 0.1241 | 3.0000 | 1.7600 | 3.8125 | 1.698c | -- | -- |
| 30 | 0.0667 | 2.0000 | 13.0000 | 0.0968 | -23.5748 | -0.0946 | -4.2700 | 6.1901 | 0.1294 | 2.0000 | 2.4800 | 3.7069 | 2.447c | 2.3327 | -0.1143 |
| 31 | 0.0645 | 2.0000 | 13.0000 | 0.0938 | 32.0889 | -0.0947 | -3.3500 | 6.1901 | 0.1511 | 2.0000 | 1.3700 | 3.7929 | 2.278c | 2.1029 | -0.1751 |
| 32 | 0.0476 | 1.0000 | 7.0000 | 0.1429 | 2.8615 | -0.0749 | -2.2300 | 3.5542 | 0.1785 | 2.0000 | 0.6400 | 3.7257 | 1.602c | 1.7672 | 0.1652 |
| 33 | 0.0588 | 2.0000 | 14.0000 | 0.0857 | 61.6262 | -0.1001 | -3.3600 | 6.4317 | 0.1432 | 2.0000 | 1.4100 | 3.9698 | 1.845c | 2.0079 | 0.1629 |
| 34 | 0.0500 | 2.0000 | 16.0000 | 0.0976 | 112.8082 | -0.1002 | -3.8000 | 7.0673 | 0.0950 | 3.0000 | 1.2300 | 4.2531 | 2.041c | 1.8581 | -0.1829 |
| 35 | 0.0571 | 2.0000 | 14.0000 | 0.0833 | -22.7851 | -0.0997 | -4.1100 | 7.0624 | 0.1164 | 3.0000 | 1.8400 | 4.1007 | 2.255c | 2.4402 | 0.1852 |
| 36 | 0.0541 | 2.0000 | 15.0000 | 0.0789 | 135.7274 | -0.1001 | -3.3700 | 6.7979 | 0.1350 | 2.0000 | 2.0600 | 4.1824 | 2.113c | 2.1202 | 0.0072 |
| 37 | 0.0556 | 2.0000 | 15.0000 | 0.0811 | 112.8082 | -0.1017 | -3.7200 | 6.2710 | 0.1224 | 2.0000 | 2.3200 | 3.8565 | 2.146c | 1.8751 | -0.2709 |
| 38d | 0.0526 | 2.0000 | 15.0000 | 0.0769 | 8.6366 | -0.1013 | -4.1900 | 7.3805 | 0.1103 | 3.0000 | 2.2200 | 4.1427 | 1.301c | -- | -- |
RNBR, Relative number of benzene rings; NBR, Number of benzene rings; NCA, Number of C atoms; RNDB, Relative number of double bonds; DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC]; MiPCN, Min partial charge for a N atom [Zefirov's PC]; ALS, Average logS; 3χ, Randic index (order 3); RPCG, RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC]; NNA, Number of N atoms; ALP, Average logP; and 2AIC, Average Information content (order 2)
Table 2.
Regression parameters and statistical quality of the correlations of the activity LogKi -CAII in the present study.
| Models | Descriptors involved | t-test | CoefficientAi (i=1,2,3,4,5) | B (intercept) | Statistical Parameters N R2 R2 cv F s2 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Equ. 1 | RNBR Relative number of benzene rings | 6.163 | 32.767(5.316) | 0.180 | 36 | 0.527 | 0.488 | 37.98 | 0.090 |
| (0.280) | |||||||||
| Equ. 2 | NBR Number of benzene rings | 7.590 | 1.296(0.170) | 1.245 | 36 | 0.647 | 0.599 | 30.27 | 0.069 |
| NCA Number of C atoms | -5.745 | -0.119(0.020) | (0.298) | ||||||
| Equ. 3 | RNDB Relative number of double bonds | -7.939 | -16.73(2.107) | 6.182 | 36 | 0.729 | 0.618 | 28.74 | 0.055 |
| DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC] | -5.458 | -0.0031(0.0005) | (0.776) | ||||||
| MiPCN Minimum partial charge for a N atom [Zefirov's PC] | 3.406 | 24.276(7.127) | |||||||
| Equ. 4 | ALS Average logS | -5.887 | -0.462(0.078) | 1.061 | 36 | 0.786 | 0.716 | 29.98 | 0.045 |
| 3χ Randic index (order 3) | -2.571 | -0.125(0.048) | (0.330) | ||||||
| RNDB Relative number of double bonds | -4.515 | -9.122(2.020) | |||||||
| RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC] | 3.924 | 7.577(1.930) | |||||||
| Equ. 5 | NBR Number of benzene rings | 7.826 | 1.739(0.222) | -2.205 | 36 | 0.840 | 0.777 | 31.54 | 0.034 |
| NCA Number of C atoms | -7.510 | -0.279(0.037) | (0.940) | ||||||
| NNA Number of N atoms | 3.578 | 0.182(0.051) | |||||||
| ALP Average logP | 3.609 | 0.282(0.078) | |||||||
| 2AIC Average Information content (order 2) | 3.488 | 0.977(0.280) | |||||||
Table 3.
Correlation matrix for the inter-correlation of various molecular descriptors involved in the obtained models.
| RNBR | NBR | NCA | RNDB | DPSA-1 | MiPCN | ALS | 3χ | RPCG | NNA | ALP | 2AIC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNBR | 1.000 | |||||||||||
| NBR | 0.6504 | 1.000 | ||||||||||
| NCA | -0.1979 | 0.5717 | 1.000 | |||||||||
| RNDB | -0.3454 | -0.3573 | 0.0337 | 1.000 | ||||||||
| DPSA-1 | -0.4510 | -0.1773 | 0.2007 | -0.4079 | 1.000 | |||||||
| MiPCN | 0.4106 | 0.1693 | -0.2183 | 0.4310 | -0.4810 | 1.000 | ||||||
| ALS | -0.2483 | -0.3005 | 0.3372 | 0.6106 | 0.5991 | -0.6935 | 1.000 | |||||
| 3χ | -0.3005 | 0.4068 | -0.2031 | -0.2953 | 0.7793 | -0.0203 | -0.7565 | 1.000 | ||||
| RPCG | 0.3372 | -0.2031 | -0.7013 | 0.2939 | 0.4134 | 0.7380 | 0.2244 | -0.5260 | 1.000 | |||
| NNA | -0.6106 | -0.2953 | 0.2939 | 0.0882 | -0.1964 | 0.3227 | -0.0431 | 0.4294 | -0.3141 | 1.000 | ||
| ALP | 0.5991 | 0.7793 | 0.4134 | -0.1964 | -0.3333 | 0.2484 | -0.7649 | 0.3105 | -0.0307 | -0.3609 | 1.000 | |
| 2AIC | -0.6935 | -0.0203 | 0.7380 | 0.3227 | 0.2484 | -0.2802 | -0.3572 | 0.7993 | -0.5658 | 0.6052 | -0.1155 | 1.000 |
RNBR, Relative number of benzene rings; NBR, Number of benzene rings; NCA, Number of C atoms; RNDB, Relative number of double bonds; DPSA-1 Difference in CPSAs (PPSA1-PNSA1) [Zefirov's PC]; MiPCN, Min partial charge for a N atom [Zefirov's PC]; ALS, Average logS; 3χ, Randic index (order 3); RPCG Relative positive charge (QMPOS/QTPLUS) [Zefirov's PC]; NNA, Number of N atoms; ALP, Average logP; and 2AIC, Average Information content (order 2).
Share and Cite
MDPI and ACS Style
Eroglu, E. Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors. Int. J. Mol. Sci. 2008, 9, 181-197. https://doi.org/10.3390/ijms9020181
AMA Style
Eroglu E. Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors. International Journal of Molecular Sciences. 2008; 9(2):181-197. https://doi.org/10.3390/ijms9020181
Chicago/Turabian StyleEroglu, Erol. 2008. "Some QSAR Studies for a Group of Sulfonamide Schiff Base as Carbonic Anhydrase CA II Inhibitors" International Journal of Molecular Sciences 9, no. 2: 181-197. https://doi.org/10.3390/ijms9020181