Synthesis of Bis (1-Methyl-2-octynyl) Ether

Instituto Universitario de Bio-Orgánica “Antonio González”, Universidad de La Laguna, Avda, Astrofísico Francisco Sánchez, 2, 38206 La Laguna, Tenerife, Spain
*
Author to whom correspondence should be addressed.
Molbank 2009, 2009(3), M612; https://doi.org/10.3390/M612
Submission received: 18 July 2009
/
Accepted: 4 August 2009
/
Published: 5 August 2009
{kind=link}
Abstract
:The synthesis of bis (1-methyl-2-octynyl) ether using two secondary alcohols under Nicholas reaction conditions is reported. The reaction is possible due to a catalytic participation of the Lewis acid when the nucleophilic alcohol is protected as a THPO-ether.
The reaction of a dicobalt octacarbonyl-stabilized propargylic cation with a nucleophile, followed by oxidative demetallation to yield propargylated products (Nicholas reaction) has been proved to be a versatile synthetic tool in organic synthesis [1,2,3,4]. This process has been used to prepare both symmetrical and unsymmetrical propargylic ethers [5], which are particularly important due to the wide range of functional group interconversions that the triple bond permits [6]. Nevertheless, when the propargylic cation is formed using a secondary alcohol, only primary alcohols are effective as nucleophiles to yield the corresponding ethers and avoid competitive elimination reactions [5].
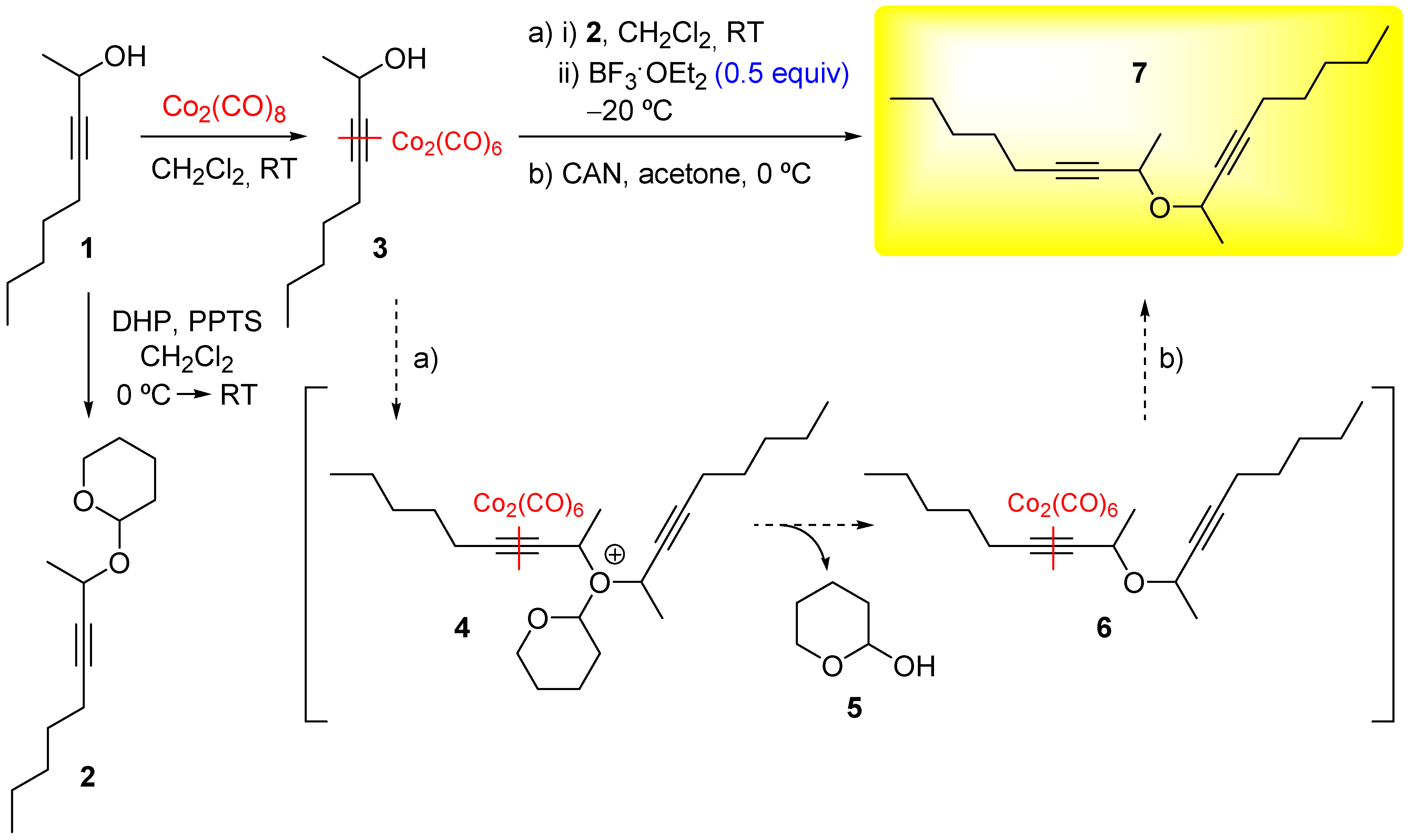
Herein, we report the synthesis of bis (1-methyl-2-octynyl) ether (7) using only the secondary alcohol 1 as electrophile and its O-protected derivative 2 as nucleophile under Nicholas reaction conditions (Scheme 1). The overall reaction is possible due to a catalytic participation of the Lewis acid when the nucleophilic alcohol is protected as a THPO-ether 2, generating the desired ether 7 and tetrahydropyran-2-ol (5) as by-product, which is in agreement with previous observations [7].
Scheme 1.
Experimental Section
General
1H and 13C NMR spectra were recorded at 25 °C on Bruker Avance-300 spectrometer in CDCl3 as solvent, and chemical shifts are reported relative to Me4Si. Low- and high-resolution mass spectra were obtained by using a Micromass Autospec spectrometer. Elemental analysis was performed on a Fisons Instrument EA 1108 CHNS-O analyzer. Infrared spectra were recorded on a Bruker IFS 55 spectrophotometer on compounds dispersed on a CaF2 disc (20 × 2 mm). Column chromatographies were performed on Merck silica gel, 60 Å and 0.2−0.5 mm. Methylene chloride was dried by distillation over calcium hydride prior to use. Compound 3-nonyn-2-ol (1) was prepared as previously described in the literature and displayed spectroscopic data identical to those reported therein [8].
Synthesis of 1-methyl-2-octynyl tetrahydro-2H-pyran-2-il ether (2)
To a stirred solution of 3-nonyn-2-ol (1) (200 mg, 1.43 mmol) in dry CH2Cl2 (8 mL) was added dihydropyran (0.20 mL, 2.15 mmol) and pyridinium p-toluenesulfonate (25 mg, 0.01 mmol) under an argon atmosphere at 0 °C. The reaction was allowed to continue at room temperature for 1 hour, after which time the mixture was poured into 20 mL of ice-water and extracted with CH2Cl2 (3 × 15 mL). The combined organic phases were washed with brine, dried (MgSO4), concentrated, and the crude purified by silica gel column chromatography yielding 2 (285 mg, 89% yield) as a yellowish oil: 1H NMR (300 MHz, CDCl3) δ/ppm = 0.87 (m, 3H), 1.29−1.31 (m, 6H), 1.41 (d, J = 7.4 Hz, 3H), 1.45−1.67 (m, 6H), 2.03 (m, 2H), 3.49−3.61 (m, 1H), 3.62−3.76 (m, 1H), 4.47−4.50 (m, 1H), 4.91 (br s, 1H); 13C NMR (75 MHz, CDCl3) δ/ppm = 13.9 (q), 18.5 (t), 19.5 (t), 22.0 (t), 22.2 (q), 25.4 (t), 28.3 (t), 30.6 (t), 33.9 (t), 61.1 (d), 62.2 (t), 81.3 (s), 85.2 (s), 98.8 (d); FT-IR (thin film) υmax (cm-1) 2875, 1458, 1310, 1115, 1091; FAB-MS m/z (relative intensity %) 224 [M]+ (11), 223 [M−1]+ (24), 153 [M−C5H11]+ (30), 85 (100). HMRS calculated for C14H24O2 [M]+ 224.177630, found 224.177120.
Synthesis of bis (1-methyl-2-octynyl) ether (7)
To a solution of alcohol 1 (100 mg, 0.71 mmol) was added dicobalt octadicarbonyl (297 mg, 0.86 mmol) in dry CH2Cl2 (7 mL) at room temperature. The reaction was stirred for 1 h, after which time the mixture was filtered through a pad of silica and the solvent evaporated to yield Co2(CO)6-propargylic ether 3 as a reddish oil. Complex 3 was dissolved in dry CH2Cl2 (7 mL) and THPO-protected propargylic alcohol 2 (302.4 mg, 1.35 mmol) was added. Then, BF3·OEt2 (29 μL, 0.23 mmol) was slowly added and the reaction mixture stirred for 1 h at −20 °C. The mixture was poured with vigorous stirring into a saturated solution of NaHCO3 (15 mL) and extracted with CH2Cl2 (2 × 15 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated to obtain the crude Co2(CO)6-propargylic ether 6 as a reddish oil, which was used in the next step without further purification. The crude 6 was dissolved in acetone (5 mL) and the mixture cooled to 0 °C. Then, Ce(NO3)6(NH4)2 (480 mg, 0.88 mmol) was added in one portion and the mixture stirred for 5 min. The reaction mixture was concentrated and the resulting residue extracted with Et2O (3 × 10 mL). The combined organic phases were dried (MgSO4), concentrated, and the residue purified by silica gel column chromatography to yield 7 (83 mg, 45% overall yield) as a yellowish oil: 1H NMR (300 MHz, CDCl3) δ/ppm = 0.88 (t, J = 11.0 Hz, 6H), 1.27−1.37 (m, 6H), 1.40 (d, J = 6.5 Hz, 6H), 1.46−1.60 (m, 6H), 2.20 (ddd, J = 7.1, 7.1, 1.9 Hz, 4H), 4.43 (m, 2H)); 13C NMR (75 MHz, CDCl3) δ/ppm = 13.9 (q), 18.7 (t), 20.0 (q), 22.2 (t), 28.3 (t), 31.0 (t), 62.8 (d), 80.2 (s), 89.0 (s); FT-IR (thin film) υmax (cm-1) 2875, 1458, 1310, 1172, 1091; FAB-MS m/z (relative intensity %) 262 [M]+ (0.1), 247 [M−CH3]+ (34), 243 (14), 221 (17), 191 [M−C5H11]+ (5), 71 (100). Elemental analysis: Calculated for C18H30O: C, 82.38; H, 11.52. Found: C, 82.51; H, 11.80.
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Supplementary File 5Supplementary File 6Acknowledgments
This research was supported by the Spanish MICINN co-financed by the European Regional Development Fund (CTQ2008-06806-C02-01/BQU), and the Canary Islands Government.
References and Notes
- Nicholas, K.M. Acc. Chem. Res. 1987, 20, 207–221.
- Green, J.R. Curr. Org. Chem. 2001, 5, 809–826.
- Teobald, B.J. Tetrahedron 2002, 58, 4133–4170.
- Díaz, D.D.; Betancort, J.M.; Martín, V.S. Synlett 2007, 343–359.
- Díaz, D.D.; Martín, V.S. Tetrahedron Lett. 2000, 41, 9993–9996.
- March, J. Advanced Organic Chemistry; John Wiley & Sons: New York, NY, USA, 1992. [Google Scholar]
- Díaz, D.D.; Martín, T.; Martín, V.S. Org. Lett. 2001, 3, 3289–3291.
- Vasil'ev, A.A. Russ. J. Org. Chem. 1994, 30, 38–41.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Díaz Díaz, D.; Martín, V.S. Synthesis of Bis (1-Methyl-2-octynyl) Ether. Molbank 2009, 2009, M612. https://doi.org/10.3390/M612
AMA Style
Díaz Díaz D, Martín VS. Synthesis of Bis (1-Methyl-2-octynyl) Ether. Molbank. 2009; 2009(3):M612. https://doi.org/10.3390/M612
Chicago/Turabian StyleDíaz Díaz, David, and Víctor S. Martín. 2009. "Synthesis of Bis (1-Methyl-2-octynyl) Ether" Molbank 2009, no. 3: M612. https://doi.org/10.3390/M612
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.