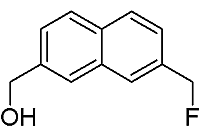
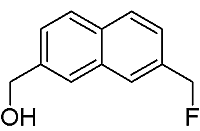
[7-(Fluoromethyl)-2-naphthyl]methanol

Institute of Chemistry, Faculty of Natural Sciences & Mathematics, Ss. Cyril and Methodius University, Arhimedova 5, 1000 Skopje, Macedonia
Molbank 2014, 2014(1), M818; https://doi.org/10.3390/M818
Submission received: 26 January 2014
/
Accepted: 21 February 2014
/
Published: 26 February 2014
Abstract
:The title compound was synthesized from 2,7-bis(bromomethyl)naphthalene via two step sequence-partial halogen exchange, followed by hydrolysis. The structure of this product was established by 1H-NMR, 13C-NMR, 19F-NMR, FT-IR spectroscopy and elemental analysis.
{kind=link}
{kind=link}
Introduction
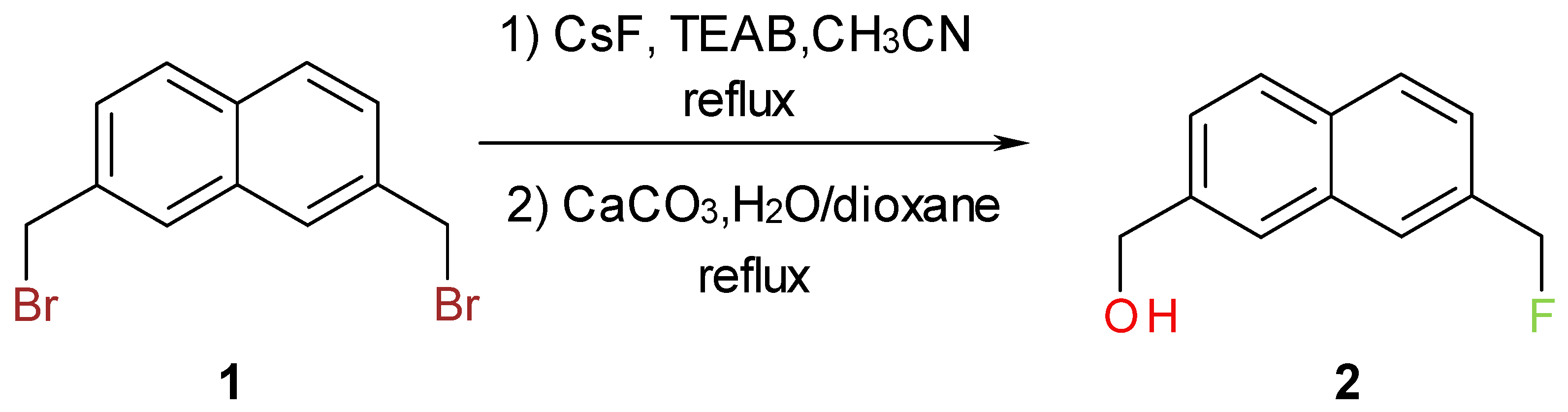
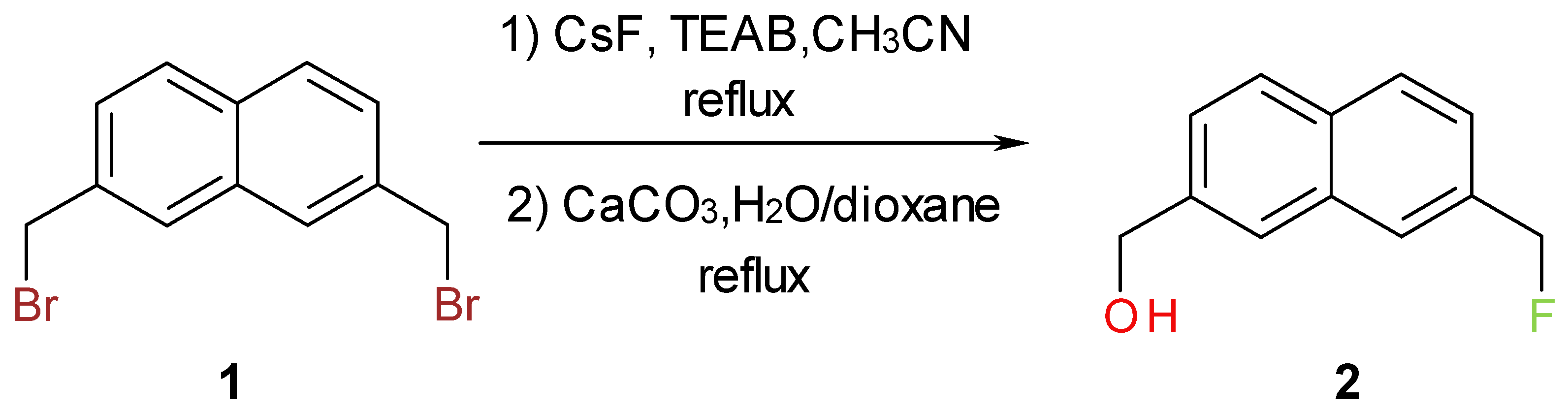
One of the usual approaches to increase the potency of biologically active compounds is the replacement of hydrogen with a fluorine atom [1,2,3]. In the course of our research on biologically active compounds, we needed to prepare bifunctional synthetic precursor, 2, containing naphthylic fluorine. The initial attempt to convert the corresponding diol, to the desired precursor using Olah’s HF/pyridine reagent [4] met with failure. The next approach was to partially hydrolyze the 2,7-bis(bromomethyl)naphthalene, 1, and to isolate the corresponding bromoalcohol, [7-(bromomethyl)-2-naphthyl]methanol [5], and then perform a halogen exchange. It was difficult to control the partial hydrolysis of 1, so the remaining approach was to subject 1 to partial halogen exchange and then to hydrolyze the 2-bromomethyl-7-fluoromethylnaphthalene. The choice of nucleophilic fluoride was cesium fluoride, because it can be conveniently dried before use and it has appreciable solubility in organic solvents [6,7,8]. Moreover, its reactivity can be conveniently enhanced by a number of useful additives, such as tetra-alkylammonium salts [9], crown ethers [10], ionic liquids [11] and bulky tert-butyl or tert-amyl alcohols [12,13]. Taking into consideration the cost, simplicity and the nature of the substrate, it was established that the most convenient procedure for preparation of pure [7-(fluoromethyl)-2-naphthyl]methanol would be by partial halogen exchange, using cesium fluoride and phase transfer catalyst in acetonitrile, followed by hydrolysis (Scheme 1).
Results and Discussion
It was very difficult to obtain 2-bromomethyl-7-fluoromethylnaphthalene in pure form because it had very similar polarity with 2,7-bis(bromomethyl)naphthalene and 2,7-bis(fluoromethyl)naphthalene. To simplify the procedure the crude mixture after partial halogen exchange was subjected to hydrolysis. The key step in this synthesis is to monitor the halogen exchange, i.e. to stop the reaction when the concentration of the 2-bromomethyl-7-fluoromethylnaphthalene is about 45%. The reaction progress can be conveniently monitored by TLC, gas chromatography (GC) or 1H-NMR. The order of elution using GC methods (HP-5 capillary columns) is: 2,7-bis(fluoromethyl)naphthalene, 2-bromomethyl-7-fluoromethylnaphthalene and 1. The title compound, 2, was prepared in two steps in 28% overall yield. The structure of 2 was unambiguously determined by spectroscopic methods (1H-NMR, 13C-NMR, 19F-NMR, FT-IR spectroscopy). The presence of benzylic fluoride is evident from the 19F-NMR spectrum (triplet centered around −207.4 ppm, arising from two bond fluorine-hydrogen coupling with coupling constant of 47.7Hz), which is supported by the 1H-NMR data (doublet centered at 5.53 ppm, 2JH-F = 47.7 Hz, 1H, Ar-CH2-F). These results are in agreement with the literature values [14,15].
Experimental Section
General
The cesium fluoride (Sigma-Aldrich, Allentown, PA, USA, 99%) was dried at 110 °C for 12 h. Acetonitrile (Sure/SealTM bottle, 99.8% anhydrous), calcium carbonate, dichloromethane and dioxane were obtained from Sigma-Aldrich and were used without further purification. Melting points were determined using Thomas-Hoover capillary mp apparatus and were uncorrected. NMR spectra were recorded on Bruker 400 MHz instrument (Freemont, CA, USA) using deuterated chloroform as solvent and tetramethylsilane as internal standard. The IR spectra were recorded on Perkin Elmer Model 1600 instrument using KBr pellets. The number of hydrogens on each carbon was determined from 13C-NMR and 135DEPT spectra. Gas chromatography was performed using a HP 6890 instrument with flame ionization detector and capillary column (HP-5, 30m, 0.32 mm i.d., 0.25 μm film thickness). Preparative flash chromatography [16] was performed using Merck silica gel 60 (230–400 mesh) TLC was carried out using Merck pre-coated plates (60 F254, 250 μm).
Synthesis of [7-(Fluoromethyl)-2-naphthyl]methanol
A mixture of 2,7-bis(bromomethyl)naphthalene[17], 1, (1.255 g, 4.00 mmol), dry cesium fluoride (1.341 g, 8.82 mmol), tetraethylammonium bromide (TEAB) (0.015 g, 0.01 mmol) in dry acetonitrile (20 mL) was vigorously stirred and refluxed under nitrogen for 24 h. The reaction progress was monitored by TLC (10% dichloromethane in hexane) and/or GC. Additional dry cesium fluoride (0.760 g, 5.00 mmol) was added and the reaction mixture was refluxed for additional 24 h. The mixture was cooled to ambient temperature, it was diluted with dichloromethane (75 mL) and transferred to a separatory funnel. The organic layer was washed with water (5 × 50 mL), brine (1 × 50 mL) and dried over sodium sulfate. Removal of solvent and drying in vacuo gave 0.923 g of off-white powder consisting of ca. 45% of the desired 2-bromomethyl-7-fluoromethylnaphthalene along with 2,7-bis(fluoromethyl)naphthalene and 2,7-bis(fluoromethyl)naphthalene. This mixture was directly used in the next step without further purification. The crude mixture was dissolved in 1:1 mixture of dioxane/water (v/v), calcium carbonate (2.8 g, 28 mmol) was added and was refluxed for 22 h. The reaction mixture was cooled to room temperature and was vacuum filtered to remove the calcium carbonate. The filter cake was washed with ethyl acetate (300 mL) and the residual calcium carbonate in the ethyl acetate was removed by a washing with 2.5% hydrochloric acid (50 mL). The organic layer was additionally washed with 5% aqueous sodium bicarbonate (1 × 50 mL), water (3 × 50 mL) and brine (1 × 50 mL). Removal of solvent and drying in vacuo resulted in 0.622 g of off-white solid, which was subjected to column chromatography (SiO2) using gradient elution (20% dichloromethane in hexane, then 10% ethyl acetate in hexane, 20% ethyl acetate in hexane, and final elution with 30% ethyl acetate in hexane). The title compound was obtained as a white crystalline solid (0.215 g, 28% overall yield).
Mp 132–134 °C (dec.).
Rf (30% ethyl acetate in hexanes) = 0.26.
GC: Rt = 16.9 min (100 °C, 3 min, 8 °C/min to 260 °C).
IR (KBr, cm−1): 3258 br. (O-H).
1H-NMR (400 MHz, CDCl3): δ = 7.87–7.81 (m, 2 H), 7.79 (s, 2H), 7.51–7.45 (m, 2 H), 5.53 (d, 2JH-F = 47.7 Hz, 1H, Ar-CH2-F), 4.84 (s, 2 H, Ar-CH2-OH), 1.93 (s, 1H, D2O exchangeable).
13C-NMR (100 MHz): δ = 139.11 (C), 134.18 (d, 2JC-F = 17 Hz, C), 133.27 (C) 133.04 (d, JC-F = 2 Hz, C), 128.50 (CH), 128.39 (CH), 126.85 (d, 3JC-F = 7 Hz, CH), 125.96 (CH), 125.74 (CH), 125.20 (d, JC-F = 5 Hz, CH), (Ar), 84.93 (d, 1JC-F = 165 Hz, Ar-CH2-F), 65.51 (Ar-CH2-OH).
135DEPT NMR: δ = 128.50 (↑, CH), 128.39 (↑, CH), 126.85 (↑, d, 3JC-F = 7 Hz, CH), 125.96 (↑, CH), 125.74 (↑, CH), 125.20 (↑, d, JC-F = 5 Hz, CH), (Ar), 84.93 (↓, d, 1JC-F = 165 Hz, Ar-CH2-F), 84.10 (↓, Ar-CH2-F), 65.51 (↓, Ar-CH2-OH).
19F NMR (CDCl3, 280 MHz): −207.4 (t, 2JH-F = 47.7 Hz, 1F, Ar-CH2-F).
Calcd. for C12H11FO, C 75.77% H 5.83%. Found: C, 75.68; H, 5.72.
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgments
The author is indebted to Alan Benesi for his help with the NMR data.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Filler, R. Organofluorine compounds in medicinal chemistry and biomedical applications. In Studies in Organic Chemistry; Filler, R., Ed.; Elsevier: New York, NY, USA, 1993; Volume 48, pp. 1–23. [Google Scholar]
- Seebach, D. Organic synthesis-Where now? Angew. Chem. Int. Ed. Engl. 1990, 29, 1320–1367. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hiyama, T. Biologically active organofluorine compounds. In Organofluorine Compounds; Springer: Berlin, Germany; Heidelberg, Germany, 2000; pp. 137–182. [Google Scholar]
- Olah, G.A.; Nojima, M.; Kerekes, I. Synthetic methods and reactions IX. Fluorination of secondary- and tertiary-alcohols with polyhydrogen fluoride/pyridine(trialkylamine) reagents. Synthesis 1973, 786–787. [Google Scholar] [CrossRef]
- Minami, A.; Uchida, R.; Eguchi, T.; Kakinuma, K. Enzymatic approach to unnatural glycosides with diverse aglycon scaffolds using glycosyltransferase VinC. J. Am. Chem. Soc. 2005, 127, 6148–6149. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H. Fluoride ion as a base in organic synthesis. Chem. Rev. 1980, 80, 429–452. [Google Scholar] [CrossRef]
- Gerstenberger, M.R.C.; Haas, A. Methods of fluorination in organic chemistry. Angew. Chem. Int. Ed. Engl. 1981, 20, 647–667. [Google Scholar] [CrossRef]
- Mascaretti, O.A. Modern methods for the monofluorination of aliphatic organic compounds. Aldrichim. Acta 1993, 26, 47–58. [Google Scholar]
- Bhadury, P.S.; Pandey, M.; Jaiswal, D.K. A facile synthesis of organofluorine compounds using a semi-molten mixture of tetrabutylammonium bromide and an alkali metal fluoride. J. Fluorine Chem. 1995, 73, 185–187. [Google Scholar] [CrossRef]
- Gingras, M.; N. Harpp, D. New anhydrous fluorinating systems: The combination of crown-ethers and cesium fluoride. A relative rate study. Tetrahedron Lett. 1988, 29, 4669–4672. [Google Scholar] [CrossRef]
- Kim, D.W.; Song, C.E.; Chi, D.Y. Significantly enhanced reactivities of the nucleophilic substitution reactions in ionic liquid. J. Org. Chem. 2003, 68, 4281–4285. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.K.; Jeong, H.J.; Seok, T.L.; Sohn, M.H.; Katzenellenbogen, J.A.; Dae, Y.C. Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: Significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J. Org. Chem. 2008, 73, 957–962. [Google Scholar]
- Shinde, S.S.; Lee, B.S.; Chi, D.Y. Synergistic effect of two solvents, tert-alcohol and ionic liquid, in one molecule in nucleophilic fluorination. Org. Lett. 2008, 10, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Dixon, E.A.; Fischer, A.; Robinson, F.P. Preparation of a series of substituted fluoromethyl-naphthalenes. Can. J. Chem. 1981, 59, 2629–2641. [Google Scholar] [CrossRef]
- Adcock, W.; Abeywickrema, A.N. Conformational preference of the fluoromethyl group in some benzyl fluorides: A 13C N.M.R. study. Aust. J. Chem. 1980, 33, 181–187. [Google Scholar] [CrossRef]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Ried, W.; Bodem, H. Characteristic derivatives of the di- and trimethylnaphthalenes. Chem. Ber. 1958, 91, 1981–1982. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of [7-(fluoromethyl)-2-naphthyl]methanol.
© 2014 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Bogdanov, J. [7-(Fluoromethyl)-2-naphthyl]methanol. Molbank 2014, 2014, M818. https://doi.org/10.3390/M818
AMA Style
Bogdanov J. [7-(Fluoromethyl)-2-naphthyl]methanol. Molbank. 2014; 2014(1):M818. https://doi.org/10.3390/M818
Chicago/Turabian StyleBogdanov, Jane. 2014. "[7-(Fluoromethyl)-2-naphthyl]methanol" Molbank 2014, no. 1: M818. https://doi.org/10.3390/M818
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.