Gold-Catalyzed Synthesis of 2-Sulfenylspiroindolenines via Spirocyclizations


Institut de Chimie des Substances Naturelles, CNRS UPR 2301, Université Paris-Sud, Université Paris-Saclay, 1 av. de la Terrasse, 91198 Gif-sur-Yvette cedex, France
*
Author to whom correspondence should be addressed.
Molbank 2018, 2018(1), M985; https://doi.org/10.3390/M985
Submission received: 7 February 2018
/
Revised: 21 February 2018
/
Accepted: 22 February 2018
/
Published: 23 February 2018
(This article belongs to the Special Issue Metal-Catalyzed Synthesis)
Abstract
:2-Phenylsulfenyl-N-propargyl tryptamines have been prepared by electrophilic sulfuration of the corresponding tryptamines. These compounds undergo gold-catalyzed spirocyclizations to the corresponding 2-phenylthiospiroindolenines in good yields. The compounds were analyzed by NMR experiments, infrared, mass spectra and X-ray diffraction. This method provides a new efficient entry to the synthesis of 2-sulfenyl spiroindolic compounds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
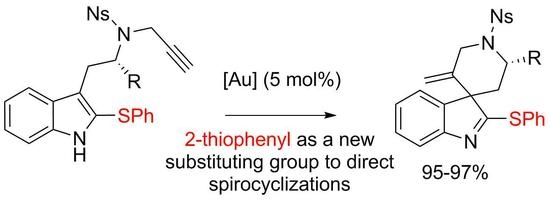
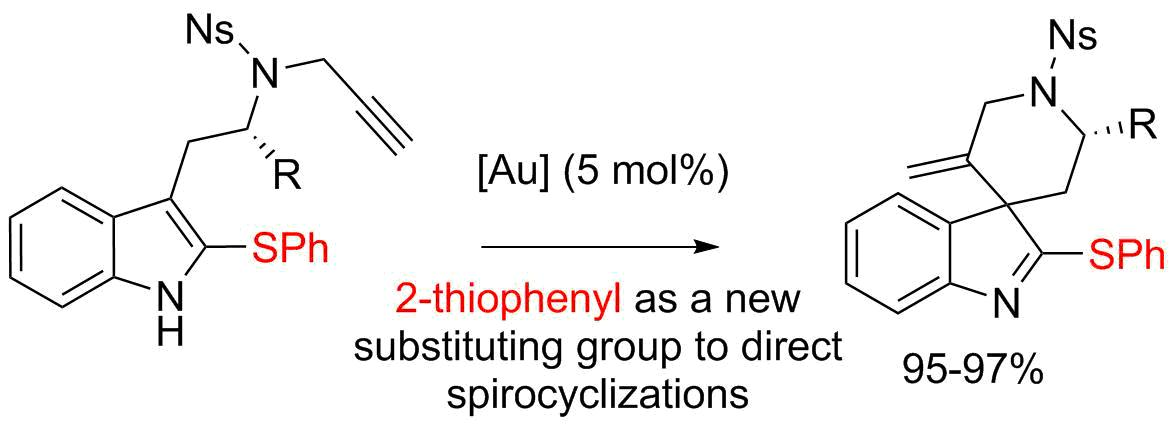
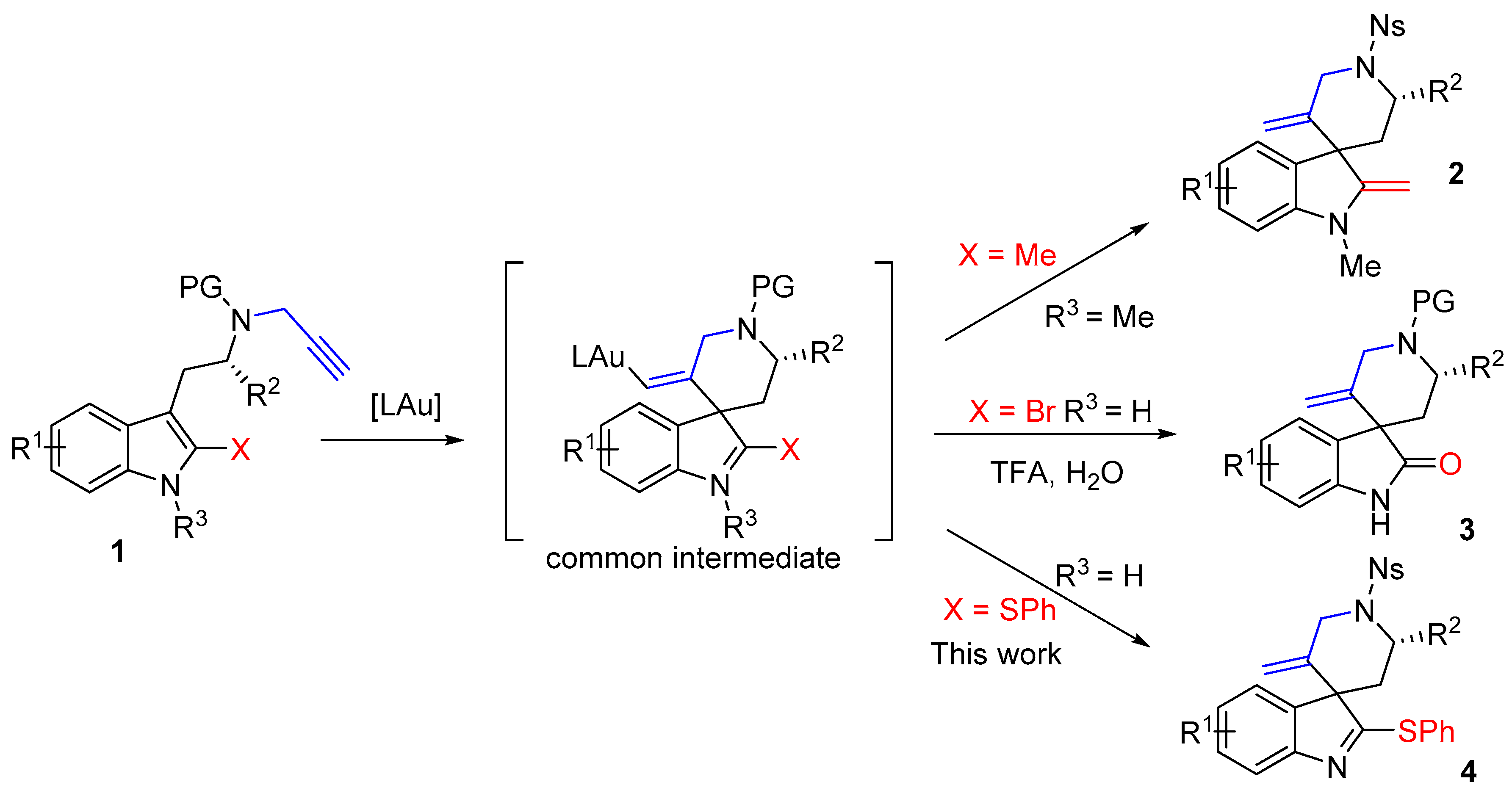
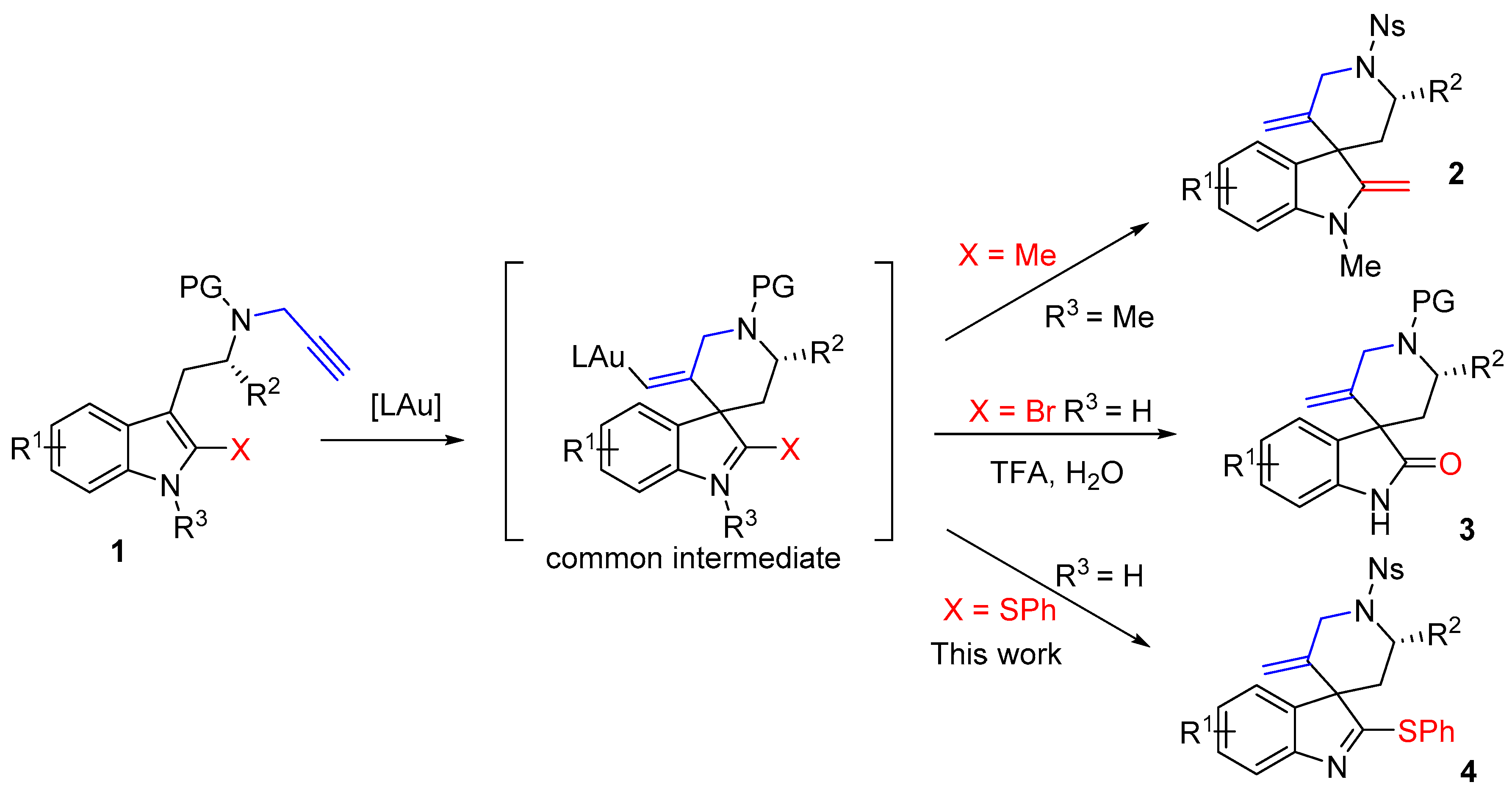
Gold complexes are highly effective catalysts to trigger the addition of nucleophiles to alkynes [1,2,3,4,5,6]. Over the last few years, the functionalization of indoles using gold catalysis has stimulated very active research in the fields of catalysis and organic synthesis. It is now well established that indoles can react with alkynes in an intramolecular fashion, upon exposure to Au(I) or Au(III) catalysts, via either their positions 2 or 3 [1,3,6]. When the alkyne is tethered to the 3-position of the indole and if position 2 is substituted, the compound will undergo dearomatizing cyclization [7,8,9], as illustrated by the conversion of the 2-methyltryptamine derivative 1 (X = Me) into the spiro-compound 2 [10] (Scheme 1). In this field, we have introduced the use of 2-bromotryptamines that led to spirooxindoles 3 after acidic hydrolysis of the spirobromoindolenine resulting from the protodeauration of the intermediate [11,12,13]. We reasoned that the scope of these spirocyclization reactions might expand to tryptamines with other substituents on their 2-positions and hypothesized that tryptamines substituted by a SPh group should lead to 2-sulfenylspiroindolenines 4. Few members of this general class of compounds were previously studied for their structural resemblance with biologically active spirooxindoles, spiroindolenines or spiroindolines [12]. Their synthetic routes were reported mainly via Pummerer reaction [14,15,16], thio-Claisen rearrangement [17], isocyanate trapping [18] or electrophilic activation upon silver catalysis [19]. In this paper we report on the use of novel 2-thiophenyltryptamines in gold(I) promoted cyclization reactions for the synthesis of original 2-thiophenylspiroindolenines.
2. Results
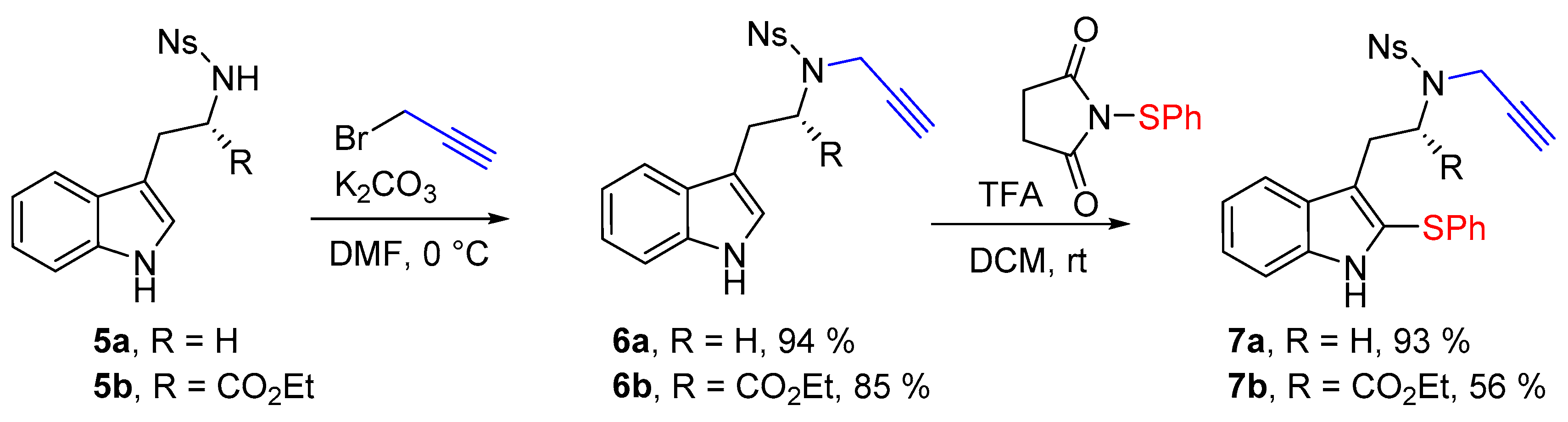
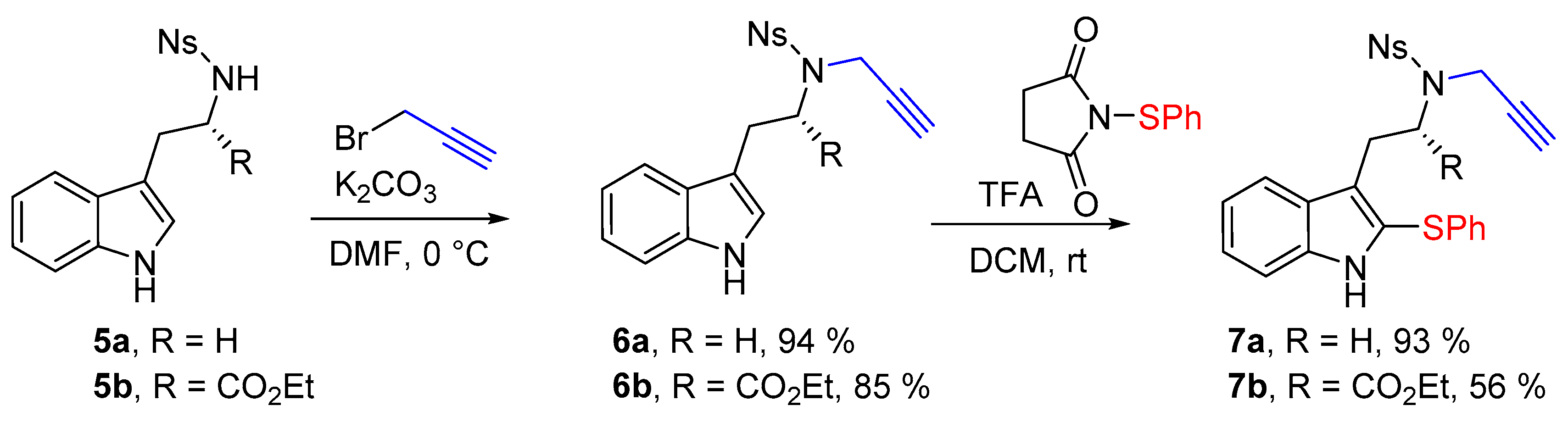
We have investigated the cyclization reactions with tryptamine and tryptophane derived substrates. Starting from N-nosyltryptamine 5a (R = H) and N-nosyltryptophane ethyl ester 5b (R = CO2Et), we have envisaged to functionalize them using the metal-free electrophilic sulfenylation reported by Cossy [20,21], because of its simplicity and efficiency. We first exposed tryptamines 5 to propargyl bromide and successfully set up the propargyl group in good yields (Scheme 2). Tryptamines 6 were next engaged in the sulfenylation reaction using N-(thiophenyl)succinimide [22] in the presence of trifluoroacetic acid. These conditions gave totally regioselective sulfenylation of the C-2 position, leading to the required tryptamine derivatives 7 in good yields.
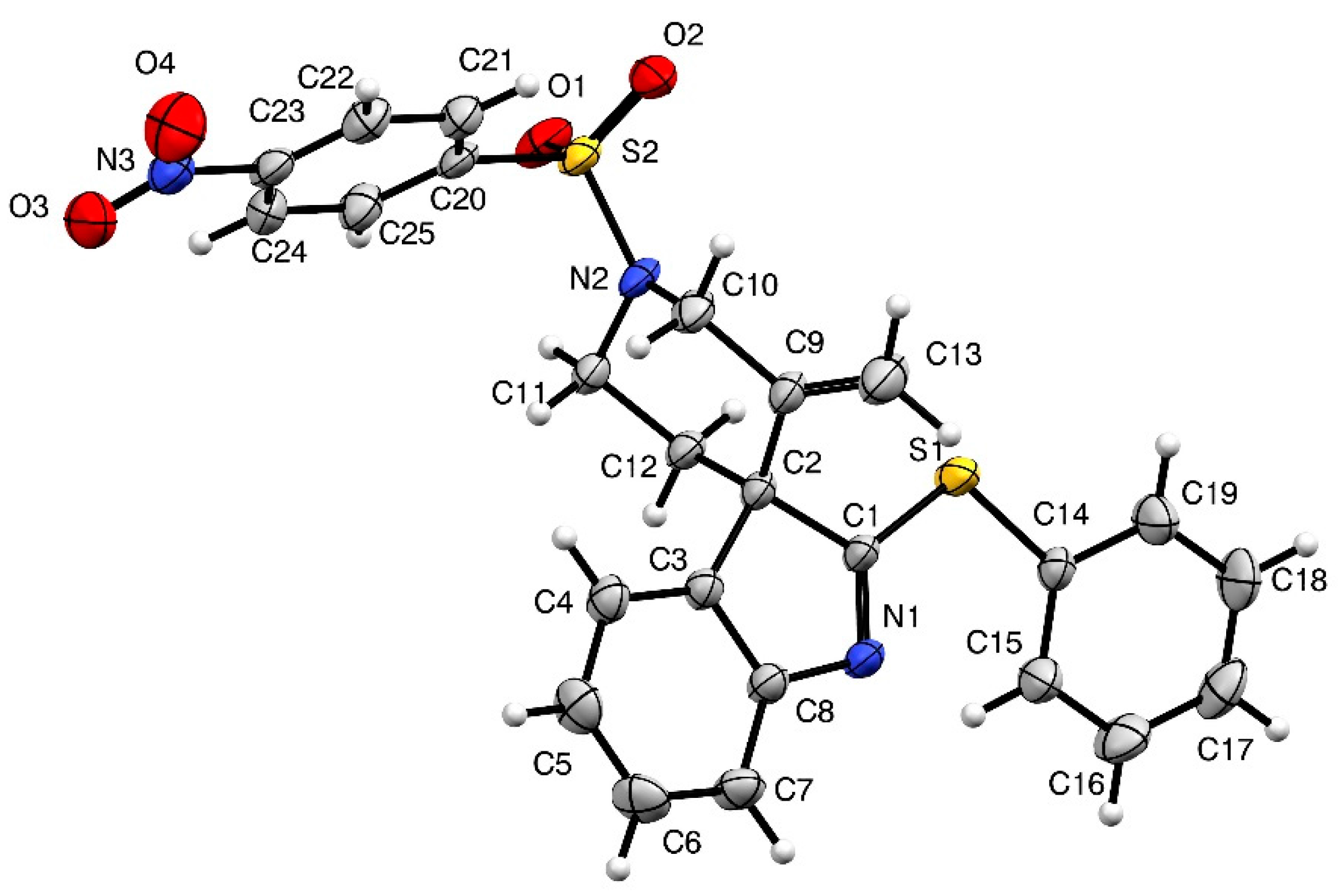
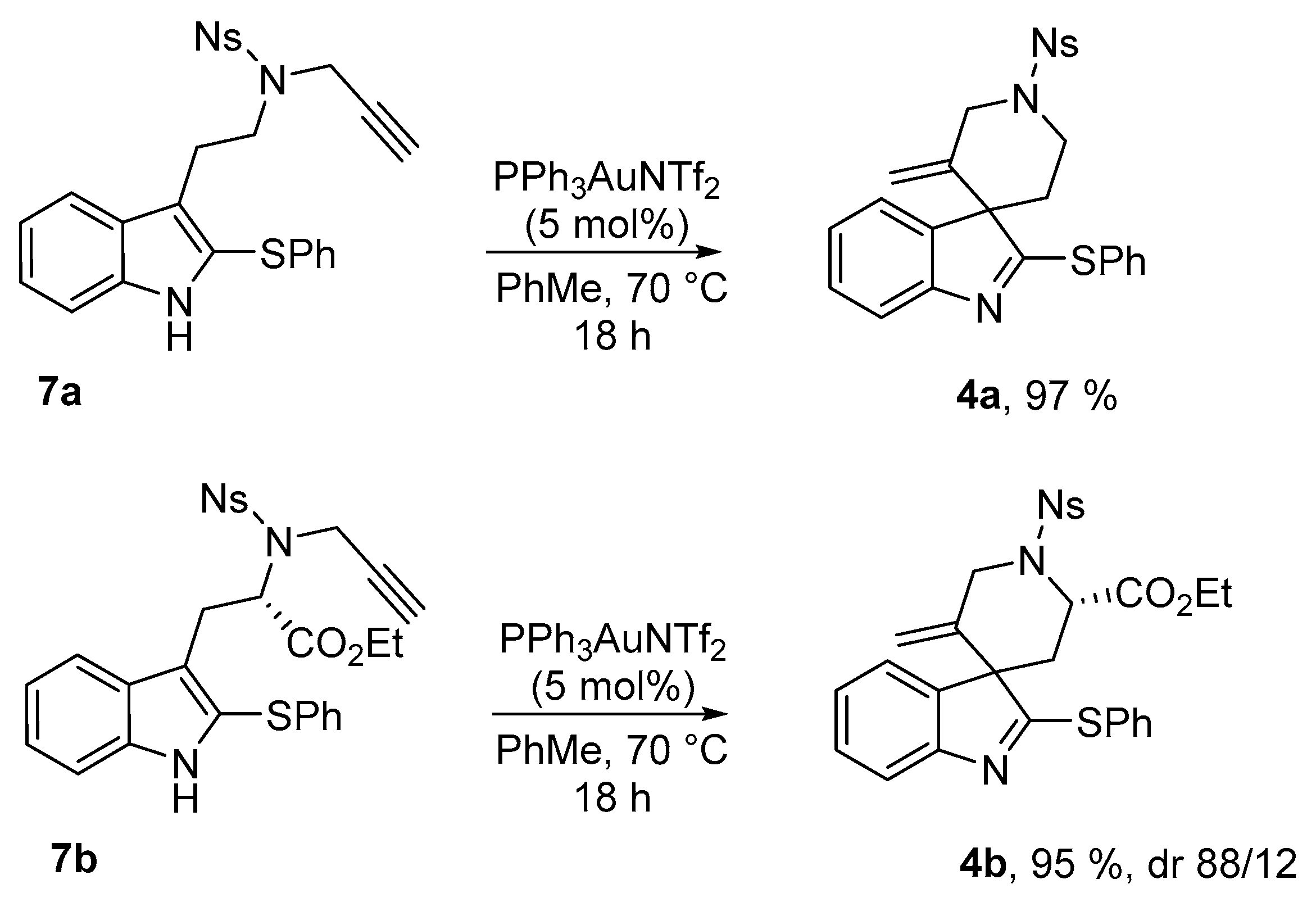
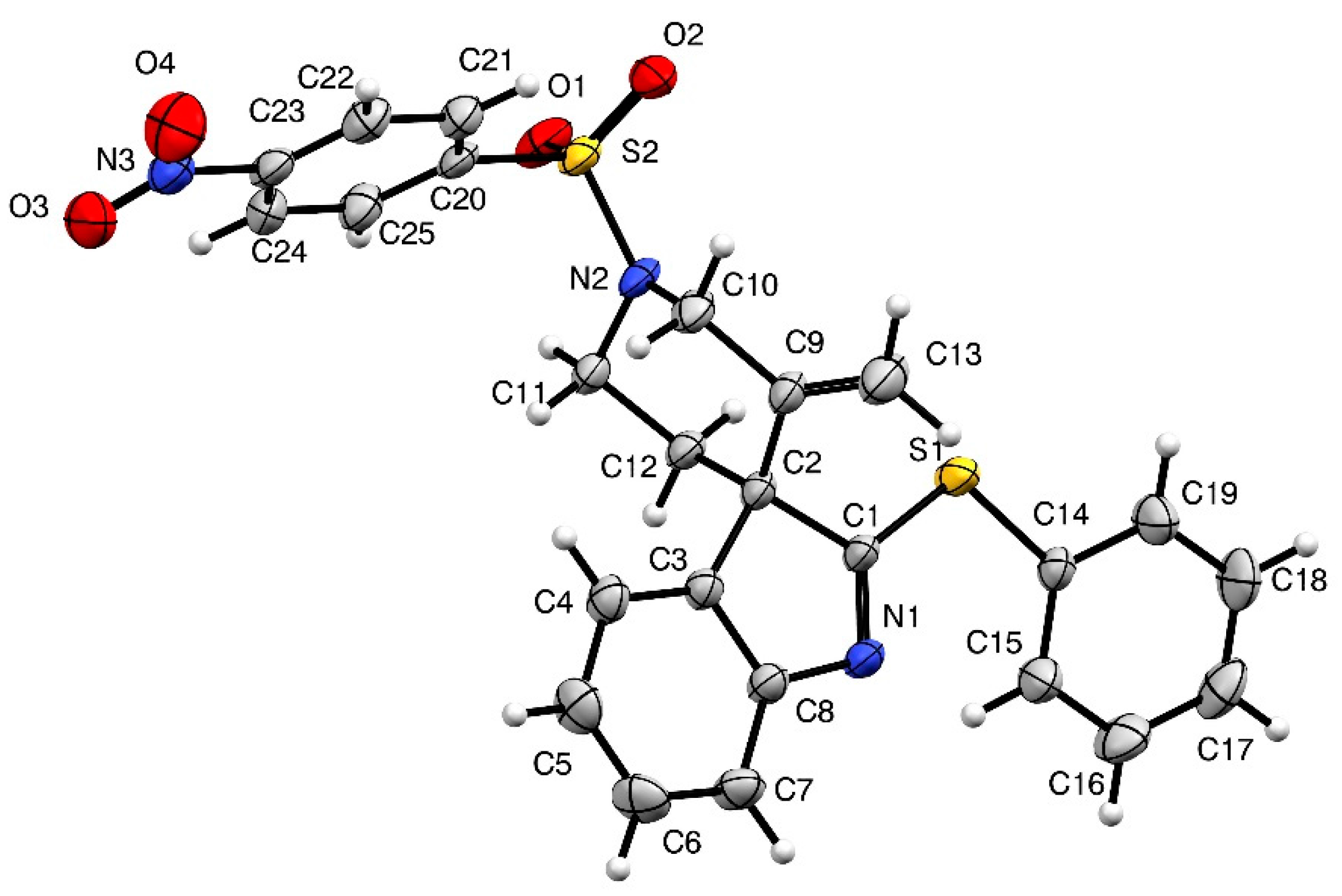
Tryptamine 7a was exposed to several Au(I) catalysts at different temperature and we finally found that the Gagosz catalyst Ph3PAuNTf2 [23] provided the best catalytic activity, leading to the desired spiroindolenine 4a in 97% isolated yield (Scheme 3). This compound showed good chemical stability and could be isolated and characterized without any trouble. Crystals of 4a suitable for X-ray diffraction experiments were grown by slow evaporation of dichloromethane and X-ray data confirmed the structural assignment. An ORTEP (Oak Ridge Thermal Ellipsoid Plot Program) drawing of 4a is displayed in Figure 1 hereafter. We pursued this study with the tryptophane derivative 7b, that led, under similar conditions, to the analogous 2-sulfenylspiroindolenine 4b in 95% yield. This compound was obtained as an inseparable 88/12 mixture of diastereomers. NOESY experiments allowed the facile determination of the relative configurations of the major diastereoisomer: major-4b displays an (S)-configuration at the quaternary carbon at the ring junction.
3. Discussion
Transition metal-catalyzed spirocyclizations of N-propargyl or N-allenyl tryptamines have been carried out so far on only a few substrates bearing substituents at the position 2 of the indole. In most of the reported examples, the indole unit is substituted by a methyl or a bromide group [10,11,12,13]. The method described herein introduces the sulfenyl group as substituent at C2 of the indole and we demonstrated its compatibility with Au(I)-catalyzed cyclization processes. This route could be applied to both tryptamine and tryptophane derivatives and provides an entry to new members of the 2-sulfenylspiroindoles series. Starting from the chiral tryptophane type substrate, gold(I)-catalyzed cyclization allowed a highly diastereoselective (dr 88/12) synthesis of the corresponding spiroindolenine. It is noteworthy that the cyclization proceeded in excellent yields for both series. Extension of the synthetic approach to spiroindolenines with 2-sulfenyl moieties has also the major advantage to allow post-functionalization of this skeleton of interest, thanks to the known reactivity of the sulfenyl group [14,15,16].
4. Materials and Methods
Reactions were performed using oven dried glasswares under an atmosphere of argon. All separations were carried out under flash-chromatographic conditions on silica gel (Redi Sep prepacked column, 230–400 mesh) at medium pressure (20 psi) with use of a CombiFlash Companion. Reactions were monitored by thin-layer chromatography on Merck silica gel plates (Merck, Darmstadt, Germany) (60 F254 aluminum sheets) which were rendered visible by ultraviolet and spraying with vanillin (15%) + sulfuric acid (2.5%) in EtOH followed by heating. CH2Cl2, DMF and toluene were purchased at the highest commercial quality and used without further purification. Reagent-grade chemicals were obtained from diverse commercial suppliers and used as received. 1H-NMR (500 or 300 MHz) and 13C-NMR (125 or 75 MHz) spectra were recorded on Bruker Avance spectrometers (Bruker, Wissembourg, France) at 298 K unless otherwise stated. Chemical shifts are given in ppm (δ) and are referenced to the internal solvent signal or to TMS used as an internal standard. Coupling constants J are given in Hz. Carbon multiplicities were determined by DEPT135 experiment. Diagnostic correlations were obtained by two-dimensional COSY, HSQC and NOESY experiments. Infrared spectra (IR) were recorded on a Perkin-Elmer FT-IR system (Waltham, MA, USA) using diamond window Dura SamplIR II and the data are reported in reciprocal centimeters (cm−1). Optical rotations were measured on a Anton Paar MCP 300 polarimeter (Anton-Paar, Courtaboeuf, France) at 589 nm. is expressed in deg·mL·g−1·dm−1 and c is expressed in g/100 mL. Melting points were recorded in open capillary tubes on a Buchi B-540 apparatus (Buchi, Rungis, France) and are uncorrected. High resolution mass spectra (HRMS) were recorded using a Micromass LCT Premier XE instrument (Waters, Milford, MA, USA) and were determined by electrospray ionization (ESI).
N-[2-(1H-Indol-3-yl)ethyl]-4-nitro-N-(prop-2-yn-1-yl)benzenesulfonamide (6a)

N-[2-(1H-Indol-3-yl)ethyl]-4-nitrobenzenesulfonamide (5a) (1.0 g, 2.9 mmol, 1 equiv.) was solubilized in dry dimethylformamide (30 mL) under argon, and K2CO3 (483 mg, 3.5 mmol, 1.2 equiv.) and propargyl bromide solution (0.31 mL, 80% w/w solution, 3.5 mmol, 1.2 equiv.) were added sequentially. The reaction mixture was stirred for 4 h at room temperature. Then a saturated aqueous solution of NH4Cl was added before pouring the reaction media in a separatory funnel containing ethyl acetate. After separation of the phases the aqueous phase was extracted twice with ethyl acetate, then the combined organic phases were washed twice with water and brine. The organic phase was then dried over MgSO4 before removing the solvent under reduced pressure. Finally the crude mixture was purified over silica gel column chromatography (eluent 10 to 50% EtOAc/Heptane), furnishing the pure product 6a as an orange foam (1.04 g, 2.71 mmol, 94%).
Rf: 0.40 (50:50 EtOAc:heptane). IR (neat) νmax: 3416, 3285, 3105, 2924, 1526, 1347, 1310, 1160 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.22 (d, J = 8.9 Hz, 2H), 8.02 (brs, 1H), 7.94 (d, J = 8.9 Hz, 2H), 7.57 (d, 7.9 Hz, 1H), 7.34 (dt, J = 8.2 and 0.9 Hz, 1H), 7.20 (ddd, J = 7.9, 7.2 and 1.1 Hz, 1H), 7.12 (ddd, J = 7.9, 7.2 and 1.1 Hz, 1H), 7.07 (d, J = 2.5 Hz, 1H), 4.23 (d, J = 2.4 Hz, 2H), 3.58 (t, J = 7.8 Hz, 2H), 3.10 (t, J = 6.7 Hz, 2H), 2.10 (t, J = 2.5 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): 150.1 (Cq), 144.9 (Cq), 136.4 (Cq), 128.9 (2CH), 127.2 (CH), 124.0 (2CH), 122.5 (CH), 122.4 (CH), 119.8 (CH), 118.6 (CH), 111.9 (Cq), 111.4 (CH), 76.4 (Cq), 74.5 (CH), 47.2 (CH2), 36.8 (CH2), 24.4 (CH2). HRMS (ESI): calcd. for C19H18N3O4S [M + H]+ 384.1018, found 384.1002.
Ethyl-N-[(4-nitrophenyl)sulfonyl]-N-(prop-2-yn-1-yl)-l-tryptophanate (6b)

Ethyl [(4-nitrophenyl)sulfonyl]-l-tryptophanate (5b) (415 mg, 1.0 mmol, 1 equiv.) was solubilized in dry dimethylformamide (10 mL) under argon, and K2CO3 (152 mg, 1.2 mmol, 1.2 equiv.) and propargyl bromide solution (0.12 mL, 80% w/w solution, 1.1 mmol, 1.1 equiv.) were added sequentially. The reaction mixture was stirred for 4 h at room temperature. Then a saturated aqueous solution of NH4Cl was added before to pour the reaction media in a separatory funnel containing ethyl acetate. After separation of the phases the aqueous phase was extracted twice with ethyl acetate, then the combined organic phases were washed twice with water and with brine. The organic phase was then dried over MgSO4 before removing the solvent under reduced pressure. Finally the crude mixture was purified over silica gel column chromatography (eluent 10 to 50% EtOAc/Heptane), furnishing the pure product 6b as an orange foam (385 mg, 0.845 mmol, 85%).
IR (neat) νmax: 3370, 3285, 3105, 3070, 1715, 1531, 1353, 1166, 1150, 1093, 887, 856, 748 cm−1. 1H-NMR (CDCl3, 300 MHz): 7.96 (brs, 1H), 7.89 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 7.5 Hz, 1H), 7.25–7.22 (m, 1H), 7.17 (td, J = 7.2 and 1.5 Hz, 1H), 7.1 (ddd, J= 7.7, 6.8 and 1.5 Hz, 1H), 6.99 (d, J = 2.5 Hz, 1H), 4.81 (dd, J = 9.6 and 5.3 Hz, 1H), 4.55 (dd, J = 18.8 and 2.5 Hz, 1H), 4.29 (dd, J = 19.8 and 2.6 Hz, 1H), 4.17 (qd, J = 7.2 and 1.7 Hz, 2H), 3.51–3.44 (m, 1H), 3.18 (dd, J = 15.0 and 9.6 Hz, 1H), 2.30 (t, J = 2.4 Hz, 1H), 1.23 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3, 75 MHz): 170.4 (Cq), 149.5 (Cq), 145.2 (Cq), 136.2 (Cq), 128.3 (2CH), 126.7 (Cq), 124.0 (CH), 123.2 (2CH), 122.5 (CH), 120.0 (CH), 118.3 (CH), 111.4 (CH), 110.0 (Cq), 79.2 (Cq), 73.4 (CH), 62.0 (CH2), 60.6 (CH), 34.2 (CH2), 26.5 (CH2), 14.2 (CH3). HRMS (ESI): calcd. for C19H18N306S [M − propargyl]− 416.0916, found 416.0908. − 15.5 (c 1.0, CHCl3).
4-Nitro-N-{2-[2-(phenylthio)-1H-indol-3-yl]ethyl}-N-(prop-2-yn-1-yl)benzenesulfonamide (7a)

To a solution of N-[2-(1H-indol-3-yl)ethyl]-4-nitro-N-(prop-2-yn-1-yl)benzenesulfonamide (6a) (383 mg, 1.0 mmol, 1 equiv.) in dry DCM (2 mL) was added 1-(phenylthio)pyrrolidine-2,5-dione (207 mg, 1.0 mmol, 1.0 equiv.), and trifluoroacetic acid (1.15 mL, 15.0 mmol, 15 equiv.). The solution was stirred for 18 h, and the crude media was quenched by a carefull addition of NaHCO3 aqueous saturated solution. The aqueous phase was then extracted two times with DCM. The combined organic phases were dried over MgSO4 and the volatiles were removed under vacuum. The crude mixture was then purified over silica gel column chromatography (eluent 20 to 50% EtOAc/Heptane), furnishing the pure desired product 7a as a yellow amorphous solid (455 mg, 0.93 mmol, 93%).
IR (neat) νmax: 3346, 3272, 3100, 3063, 1603, 1581, 1524, 1444, 1344, 1160, 1090, 855 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.14 (d, J = 9.2 Hz, 2H), 8.08 (brs, 1H), 7.84 (d, J = 9.2 Hz, 2H), 7.61 (d, J = 7.7 Hz, 1H), 7.29–7.27 (m, 1H), 7.25-7.14 (m, 5H), 7.06-7.03 (m, 2H), 4.26 (d, J = 2.5 Hz, 2H), 3.49 (dd, J = 9.0 and 6.3 Hz, 2H), 3.17 (dd, J = 9.1 and 6.8 Hz, 2H), 2.05 (t, J = 2.5 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): 147.2 (Cq), 144.9 (Cq), 137.1 (Cq), 136.8 (Cq), 129.4 (2CH), 128.8 (2CH), 127.5 (Cq), 126.9 (2CH), 126.3 (CH), 124.0 (CH), 124.0 (2CH), 123.0 (Cq), 120.4 (CH), 119.4 (Cq), 119.2 (CH), 111.3 (CH), 76.4 (Cq), 74.5 (CH), 46.9 (CH2), 36.8 (CH2), 24.0 (Cq). HRMS (ESI): calcd. for C25H22N3O4S2 [M + H]+ 492.1046, found 492.1053.
Ethyl-(S)-2-[(4-nitro-N-(prop-2-yn-1-yl)phenyl)sulfonamido)-3-[2-(phenylthio)-1H-indol-3-yl]propanoate (7b)

To solution of ethyl N-[(4-nitrophenyl)sulfonyl]-N-(prop-2-yn-1-yl)-l-tryptophanate (6b) (385 mg, 0.85 mmol, 1 equiv.) in dry DCM (2 mL) was added 1-(phenylthio)pyrrolidine-2,5-dione (184 mg, 0.89 mmol, 1.05 equiv.), and trifluoroacetic acid (0.98 mL, 12.75 mmol, 15 equiv.). The solution was stirred for 18 h, and the crude media was quenched by a careful addition of NaHCO3 aqueous saturated solution. The aqueous phase was then extracted two times with DCM. The combined organic phases were dried over MgSO4 and the volatiles were removed under vacuum. The crude mixture was then purified over silica gel column chromatography (eluent 20 to 50% EtOAc/Heptane), furnishing the pure desired product 7b as a yellow amorphous solid (269 mg, 0.477 mmol, 56%).
IR (neat) νmax: 3372, 3287, 3105, 3070, 2926, 1734, 1717, 1530, 1349, 1164, 1093, 855, 739 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.07 (brs, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.56 (dd, J = 7.9 and 0.9 Hz, 1H), 7.25–7.10 (m, 6H), 7.03–7.00 (m, 2H), 4.91 (t, J = 7.8 Hz, 1H), 4.46 (t, J = 2.2 Hz, 2H), 3.99 (q, J = 7.2 Hz, 2H), 3.57 (dd, J = 14.5 and 7.5 Hz, 1H), 3.31 (dd, J 14.5 and 8.1 Hz, 1H), 2.21 (t, J = 2.5 Hz, 1H), 1.04 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3, 75 MHz): 170.1 (Cq), 149.8 (Cq), 145.6 (Cq), 137.0 (Cq), 136.3 (Cq), 129.5 (2CH), 128.7 (2CH), 127.4 (Cq), 127.0 (2CH), 126.4 (CH), 124.1 (CH), 123.6 (2CH + Cq), 120.5 (CH), 119.2 (CH), 117.5 (Cq), 111.3 (CH), 78.7 (Cq), 73.7 (CH), 61.9 (CH2), 59.7 (CH), 34.4 (CH2), 26.1 (CH2), 13.9 (CH3). HRMS (ESI): calcd. for C28H26N3O6S2 [M + H]+ 564.1258, found 464.1268. − 50.4 (c 1.0 CHCl3).
3′-Methylene-1′-[(4-nitrophenyl)sulfonyl]-2-(phenylthio)spiro[indole-3,4′-piperidine] (4a)

4-Nitro-N-{2-[2-(phenylthio)-1H-indol-3-yl]ethyl}-N-(prop-2-yn-1-yl)benzenesulfonamide (7a) (116 mg, 0.236 mmol, 1 equiv.) was pourred in a shlenck tube filled with argon, dry toluene (2.4 mL) and PPh3AuNTf2 (8.7 mg, 0.012 mmol, 5 mol %) were added, and the solution was stirred at 70 °C for 15 h. Then the solvent was evaporated and the crude mixture was purified over silica gel column chromatography (10 to 30% EtOAc:Heptane), furnishing the pure product 4a as a yellowish solid (113 mg, 0.229 mmol, 97%).
IR (neat) νmax: 3347, 3273, 3104, 2920, 1607, 1586, 1527, 1516, 1348, 1314, 1166, 1148, 933, 855, 742 cm−1. Mp 223 °C. 1H-NMR (CDCl3, 500 MHz): 8.46 (d, J = 8.9 Hz, 2H), 8.10 (d, J = 9.0 Hz, 2H), 7.57–7.55 (m, 2H), 7.43–7.41 (m, 3H), 7.38 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 7.8 Hz, 1H), 5.07 (s, 1H), 4.78 (s, 1H), 4.29 (d, J = 14.3 Hz, 1H), 4.21 (d, J = 14.1 Hz, 1H), 3.84–3.79 (m, 1H), 3.71–3.66 (m, 1H), 2.11–2.06 (m, 1H), 1.93–1.88 (m, 1H). 13C-NMR (CDCl3, 75 MHz): 182.6 (Cq), 154.0 (Cq), 150.5 (Cq), 143.7 (Cq), 142.9 (Cq), 137.5 (Cq), 134.4 (2CH), 129.6 (CH), 129.5 (2CH), 129.0 (2CH), 128.7 (Cq), 128.0 (Cq), 124.8 (CH), 124.7 (2CH), 122.3 (CH), 120.4 (CH), 116.0 (CH2), 62.6 (Cq), 50.2 (CH2), 42.5 (CH2), 33.9 (CH2). HRMS (ESI): calcd. C25H22N3O4S2 [M + H]+ 492.1046, found 492.1052.
Ethyl-(2′S,3S)-5′-methylene-1′-[(4-nitrophenyl)sulfonyl]-2-(phenylthio)spiro[indole-3,4′-piperidine]-2′-carboxylate (4b)

Ethyl-(S)-2-{[4-nitro-N-(prop-2-yn-1-yl)phenyl]sulfonamide}-3-[2-(phenylthio)-1H-indol-3-yl]propanoate (7b) (56.4 mg, 0.1 mmol, 1 equiv.) was pourred in a shlenck tube filled with argon, dry toluene (1 mL) and PPh3AuNTf2 (3.7 mg, 0.005 mmol, 5 mol %) were added, and the solution was stirred at 70 °C for 15 h. Then the solvent was evaporated and the crude mixture was purified over silica gel column chromatography (10 to 30% EtOAc:Heptane), furnishing the pure product 4b as a yellowish solid (53.7 mg, 0.95 mmol, 95%). A 88/12 mixture of diastereoisomers was obtained and was characterized without further separation. For 1H-NMR only the most abundant diastereomer is described.
IR (neat) νmax: 3105, 3065, 2983, 1740, 1606, 1529, 1454, 1348, 1310, 1164, 1023, 909, 732 cm−1. Mp 82–83 °C. 1H-NMR of major diastereoisomer (CDCl3, 500 MHz): 8.41 (d, J = 8.9 Hz, 2H), 8.15 (d, J = 8.9 Hz, 2H), 7.62–7.60 (m, 2H), 7.45–7.41 (m, 4H), 7.29 (td, J = 7.5 and 1.2 Hz, 1H), 7.18–7.16 (m, 1H), 7.09 (td, J = 7.4 and 0.8 Hz, 1H), 5.12 (s, 1H), 4.83 (dd, J = 11.1 and 5.9 Hz, 1H), 4.60 (s, 1H), 4.47 (d, J = 15.3 Hz, 1H), 4.28 (dt, J = 15.2 and 1.8 Hz, 1H), 4.19-4.07 (m, 2H), 2.63 (dd, J = 14.2 and 11.0 Hz, 1H), 2.08 (dd, J = 14.2 and 6.0 Hz, 1H), 1.24 (t, J = 7.2 Hz, 3H). 13C-NMR of major diastereoisomer (CDCl3, 125 MHz): 183.6 (Cq), 171.0 (Cq), 154.4 (Cq), 150.3 (Cq), 145.6 (Cq), 144.0 (Cq), 138.9 (Cq), 134.4 (2CH), 129.6 (CH), 129.6 (2CH), 129.1 (2CH), 128.8 (CH), 127.7 (Cq), 125.4 (CH), 124.4 (2CH), 122.4 (CH), 120.3 (CH), 115.7 (CH2), 62.3 (CH2), 61.3 (Cq), 53.9 (CH), 47.6 (CH2), 35.1 (CH2), 14.2 (CH3). 13C-NMR of minor diastereoisomer (CDCl3, 125 MHz): 183.3 (Cq), 170.8 (Cq), 153.5 (Cq), 145.6 (Cq), 143.5 (Cq), 137.3 (Cq), 134.7 (Cq), 134.4 (2CH), 129.0 (2CH), 128.7 (CH), 127.9 (Cq), 124.7 (CH), 124.4 (2CH), 121.9 (CH), 111.5 (CH2), 62.3 (CH2), 61.9 (Cq), 53.7 (CH), 47.5 (CH2), 35.9 (CH2), 14.0 (CH3). HRMS (ESI): calcd. for C28H26N3O6S2 [M + H]+ 564.1258, found 464.1254.
5. Conclusions
In conclusion, we have reported a new efficient route to 2-sulfenylspiroindolenines using a gold-catalyzed spirocyclization as the key reaction. The key point in this field is often the nature of the substituent at position 2 of the indole, allowing either the synthesis of spiro compounds, or achiral compounds if position 2 is unsubstituted. The synthetic strategy developed features the use of a thiophenyl substituting group at the position 2 of the indole that serves as a directing group to ensure a full regioselectivity at position 3 of the indole during the cyclization. The spiranic derivatives were obtained in excellent yields (>95%) and diastereoselectivity (88/12 dr) when an N-propargyl tryptophane was used. With the ease and excellent regioselectivity of the sulfenylation of the indole and efficiency of spirocyclization, this method might pave the way to the design of new spirocyclizations of indole derivatives.
Supplementary Materials
The following are available online, NMR spectra for all new compounds, cif file for 4a.
Acknowledgments
V.M. thanks the ICSN for financial support.
Author Contributions
V.M. performed the experiments and analyzed the IR, MS, and NMR spectral data. P.R. performed the XRay analyses. A.M., A.V. and X.G. conceived and designed the project. X.G. wrote the paper.
References
- Bandini, M. Gold-catalyzed decorations of arenes and heteroarenes with C-C multiple bonds. Chem. Soc. Rev. 2011, 40, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Hashmi, A.S.K. Heterocycles from gold catalysis. Chem. Commun. 2011, 47, 6536–6544. [Google Scholar] [CrossRef] [PubMed]
- Britton, J.; Camp, J.E. Synthesis of heterocycles via gold multifaceted catalysis. Chem. Today 2012, 30, 6–8. [Google Scholar]
- Huang, H.; Zhou, Y.; Liu, H. Recent advances in the gold-catalyzed additions to C–C multiple bonds. Beilstein J. Org. Chem. 2011, 7, 897–936. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Núñez, E.; Echavarren, A.M. Molecular diversity through gold catalysis with alkynes. Chem. Commun. 2007, 333–346. [Google Scholar] [CrossRef]
- Magné, V.; Marinetti, A.; Gandon, V.; Voituriez, A.; Guinchard, X. Synthesis of Spiroindolenines via Regioselective Gold(I)-Catalyzed Cyclizations of N-Propargyl Tryptamines. Adv. Synth. Catal. 2017, 359, 4036–4042. [Google Scholar] [CrossRef]
- Roche, S.P.; Youte Tendoung, J.J.; Tréguier, B. Advances in dearomatization strategies of indoles. Tetrahedron 2015, 71, 3549–3591. [Google Scholar] [CrossRef]
- Denizot, N.; Tomakinian, T.; Beaud, R.; Kouklovsky, C.; Vincent, G. Synthesis of 3-arylated indolines from dearomatization of indoles. Tetrahedron Lett. 2015, 56, 4413–4429. [Google Scholar] [CrossRef]
- Roche, S.P.; Porco, J.A. Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem. Int. Ed. 2011, 50, 4068–4093. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.; Amijs, C.H.M.; Echavarren, A.M. Intra- and Intermolecular Reactions of Indoles with Alkynes Catalyzed by Gold. Chem. Eur. J. 2007, 13, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Magné, V.; Blanchard, F.; Marinetti, A.; Voituriez, A.; Guinchard, X. Synthesis of Spiro[piperidine-3,3′-oxindoles] via Gold(I)-Catalyzed Dearomatization of N-Propargyl- and N-Homoallenyl-2-bromotryptamines. Adv. Synth. Catal. 2016, 358, 3355–3361. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.Q.; Liu, H.M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Liddon, J.T.R.; Clarke, A.K.; Taylor, R.J.K.; Unsworth, W.P. Preparation and Reactions of Indoleninyl Halides: Scaffolds for the Synthesis of Spirocyclic Indole Derivatives. Org. Lett. 2016, 18, 6328–6331. [Google Scholar] [CrossRef] [PubMed]
- Feldman, K.S.; Boneva Vidulova, D. Use of Stang’s reagent, PhI(CN)OTf, to promote Pummerer-like oxidative cyclization of 2-(phenylthio)indoles. Tetrahedron Lett. 2004, 45, 5035–5037. [Google Scholar] [CrossRef]
- Feldman, K.S.; Vidulova, D.B. Extending Pummerer Reaction Chemistry. Application to the Oxidative Cyclization of Indole Derivatives. Org. Lett. 2004, 6, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Feldman, K.S.; Vidulova, D.B.; Karatjas, A.G. Extending Pummerer Reaction Chemistry. Development of a Strategy for the Regio- and Stereoselective Oxidative Cyclization of 3-(ω-Nucleophile)-Tethered Indoles. J. Org. Chem. 2005, 70, 6429–6440. [Google Scholar] [CrossRef] [PubMed]
- Novikov, A.V.; Kennedy, A.R.; Rainier, J.D. Sulfur Ylide-Initiated Thio-Claisen Rearrangements. The Synthesis of Highly Substituted Indolines. J. Org. Chem. 2003, 68, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Cochard, F.; Sapi, J.; Laronze, J.Y. A novel and convenient access to highly substituted spiro[pyrrolidinon-3,3′-indoles]. Tetrahedron Lett. 2001, 42, 6291–6294. [Google Scholar] [CrossRef]
- Liddon, J.T.R.; James, M.J.; Clarke, A.K.; O’Brien, P.; Taylor, R.J.K.; Unsworth, W.P. Catalyst-Driven Scaffold Diversity: Selective Synthesis of Spirocycles, Carbazoles and Quinolines from Indolyl Ynones. Chem. Eur. J. 2016, 22, 8777–8780. [Google Scholar] [CrossRef] [PubMed]
- Hostier, T.; Ferey, V.; Ricci, G.; Gomez Pardo, D.; Cossy, J. Synthesis of Aryl Sulfides: Metal-Free C–H Sulfenylation of Electron-Rich Arenes. Org. Lett. 2015, 17, 3898–3901. [Google Scholar] [CrossRef] [PubMed]
- Hostier, T.; Ferey, V.; Ricci, G.; Gomez Pardo, D.; Cossy, J. TFA-promoted direct C-H sulfenylation at the C2 position of non-protected indoles. Chem. Commun. 2015, 51, 13898–13901. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, P.; Anbarasan, P. Palladium Catalyzed Aryl(alkyl)thiolation of Unactivated Arenes. Org. Lett. 2014, 16, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Mézailles, N.; Ricard, L.; Gagosz, F. Phosphine Gold(I) Bis-(trifluoromethanesulfonyl)imidate Complexes as New Highly Efficient and Air-Stable Catalysts for the Cycloisomerization of Enynes. Org. Lett. 2005, 7, 4133–4136. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Spirocyclizations of N-propargyl tryptamines.
Scheme 2.
Synthesis of substrates 7a,b.
Scheme 3.
Au(I)-catalyzed spirocyclizations of tryptamines 7.
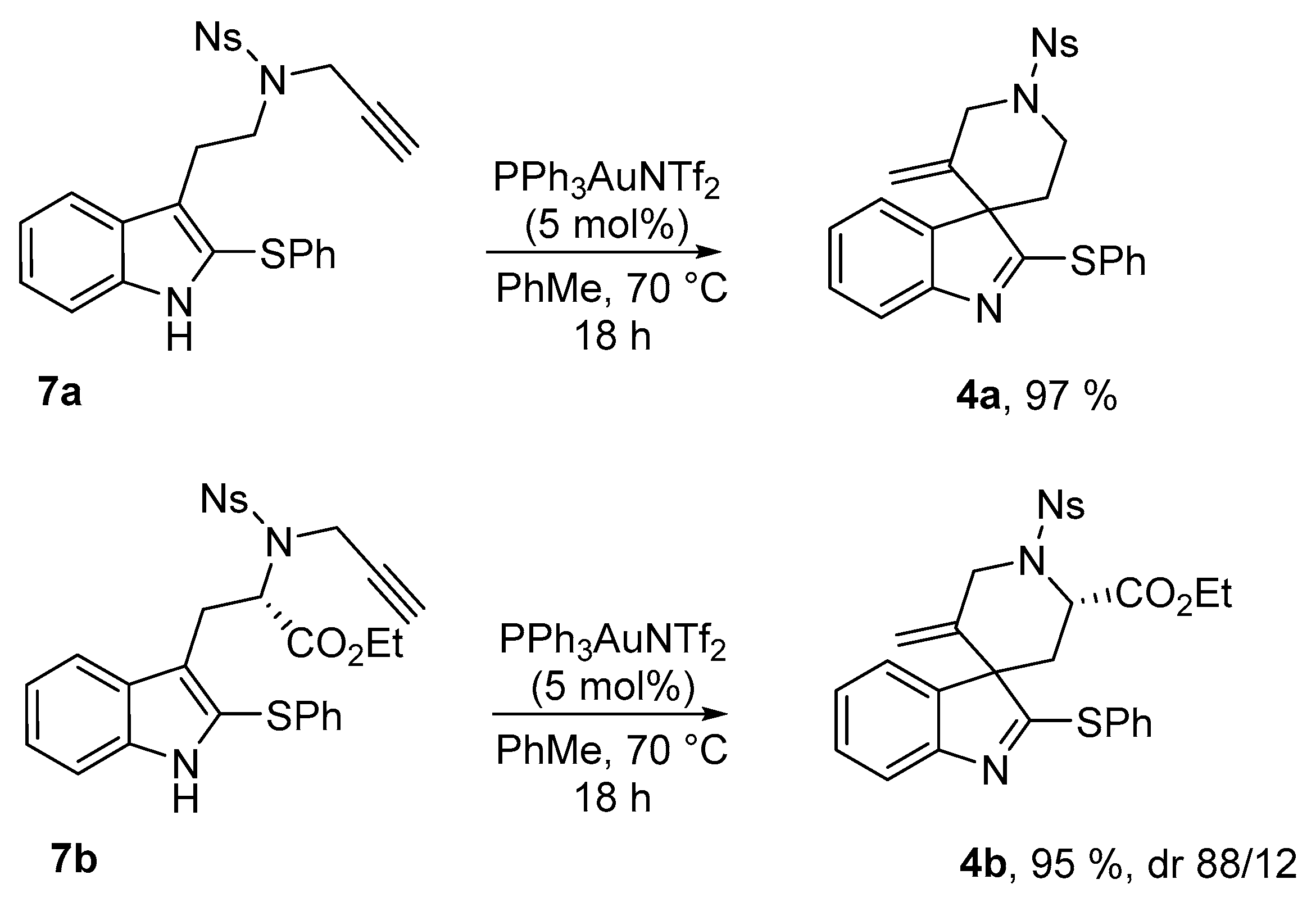
Figure 1.
Structure of compound 4a (thermal ellipsoids set at 50% probability).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Magné, V.; Retailleau, P.; Marinetti, A.; Voituriez, A.; Guinchard, X. Gold-Catalyzed Synthesis of 2-Sulfenylspiroindolenines via Spirocyclizations. Molbank 2018, 2018, M985. https://doi.org/10.3390/M985
AMA Style
Magné V, Retailleau P, Marinetti A, Voituriez A, Guinchard X. Gold-Catalyzed Synthesis of 2-Sulfenylspiroindolenines via Spirocyclizations. Molbank. 2018; 2018(1):M985. https://doi.org/10.3390/M985
Chicago/Turabian StyleMagné, Valentin, Pascal Retailleau, Angela Marinetti, Arnaud Voituriez, and Xavier Guinchard. 2018. "Gold-Catalyzed Synthesis of 2-Sulfenylspiroindolenines via Spirocyclizations" Molbank 2018, no. 1: M985. https://doi.org/10.3390/M985
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.