3.2. Phylogenetic Reconstructions, Amino Acid Sequences and Recombination Analysis
Forty-one
Xaj strains obtained from different cultivars and geographic areas of walnut cultivation and four strains obtained from walnut fruit apical necrosis were typed at four loci,
acnB,
gapA,
gyrB and
rpoD. Each locus provided between 589 and 775 bp of common sequence resulting in a total sequence length of 2,556 bp. A ML dendrogram was constructed using either sequences of each locus or concatenated data of the four genes. The best-fit nucleotide substitution model used to infer the ML dendrogram was consistent among the four genes and fit in the GTR (general time-reversible) model. In each case, both the hLRT and AIC found the same model. Either the dendrogram built with ML or the radial dendrogram obtained with NJ algorithms showed the same phylotype distribution among the
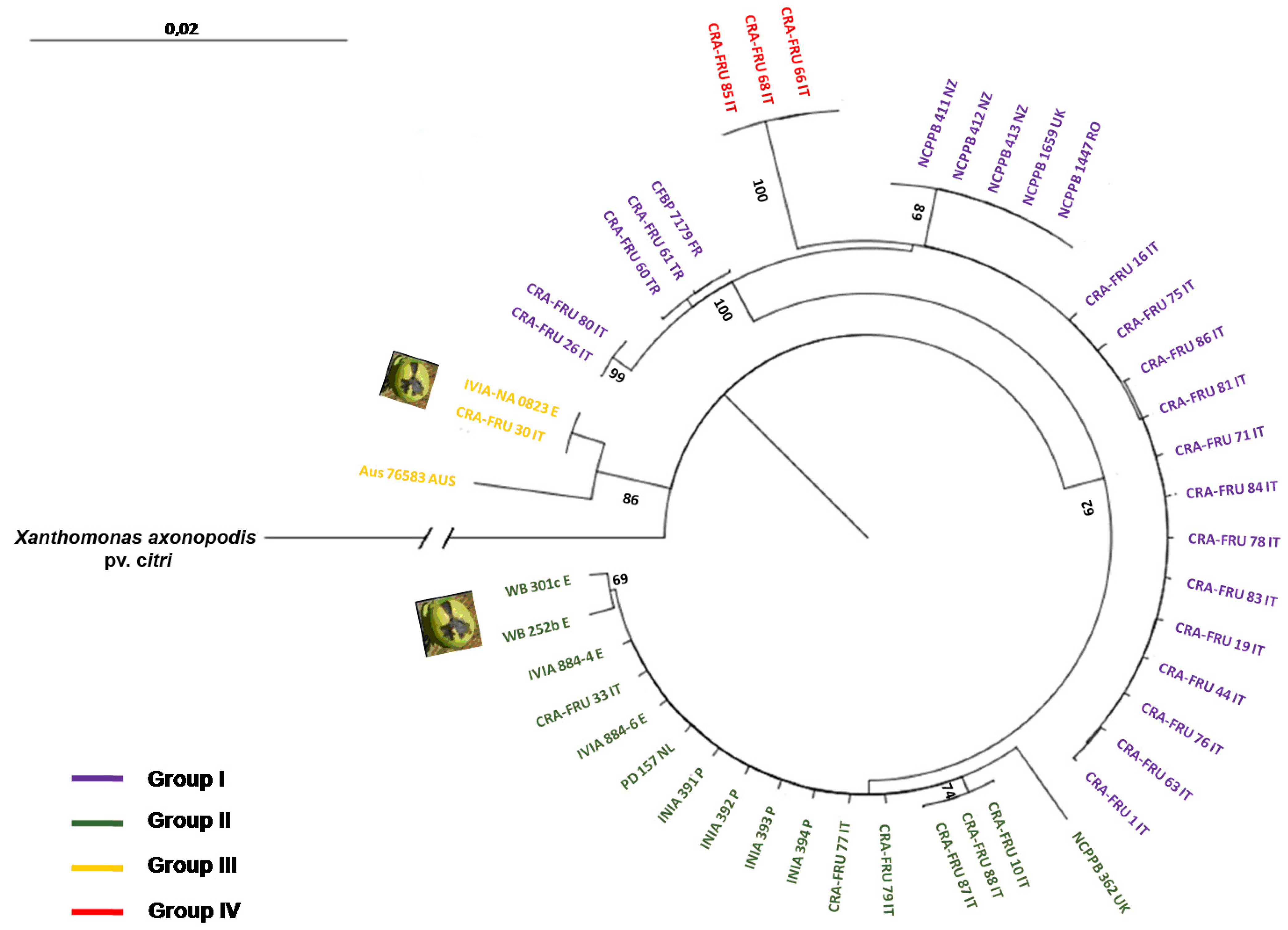
Xaj strains. The radial dendrogram using concatenated data and the NJ algorithm is shown in
Figure 3. It reveals two main phylotypes within
Xaj strains, termed groups I and II. In addition, two other minor phylotypes, named groups III and IV, were also found. Phylotype I contains a subgroup with the type-strain of the pathovar (
i.e., NCPPB 411), two others strains from New Zealand, one strain from the United Kingdom and one strain from Romania, all showing the same gene sequences. This phylotype also includes a subgroup of 15 homogenous
Xaj strains isolated in 2006 and 2008 in Rome and Caserta, Italy, as well as a subgroup with two strains obtained from Turkey and one from France showing the same gene sequences. Phylotype II includes a main subgroup with strains isolated from Italy, Spain, Portugal and the Netherlands all showing the same sequences. This phylotype also contains two
Xaj strains obtained in Spain from fruit apical necrosis. Phylotype III contains two strains isolated in Italy and Spain from walnut apical necrosis, which showed the same sequences, and one strain from Australia. Phylotype IV includes three
Xaj strains, isolated in 2008 in Rome, Italy, which showed a triplet deletion in the
rpoD gene fragment. The strains of this phylotype are the same as phylotype I, except for a mutation within the
rpoD gene fragment. The sequences diversity of the four
Xaj phylotypes was relatively high, varying from 0.567 (phylotype I) to 0.667 (phylotype III). The
Xaj strains isolated in Caserta, Italy, showed the highest level of haplotypic diversity (
Table 3).
Table 3.
Genetic diversity observed within Xanthomonas arboricola pv. juglandis according to phylogenetic reconstruction (phylotypes) and geographic origin of the strains.
Table 3.
Genetic diversity observed within Xanthomonas arboricola pv. juglandis according to phylogenetic reconstruction (phylotypes) and geographic origin of the strains.
| Phylotype/Origin of strains | Number of sequences | Number of haplotypes | Number of polymorphic site | Haplotype diversity |
|---|
| I | 23 | 4 | 16 | 0.636 |
| II | 16 | 5 | 33 | 0.600 |
| III | 3 | 2 | 25 | 0.667 |
| IV | 3 | 1 | 0 | 0.000 |
| Italy | 25 | 6 | 57 | 0.707 |
| Italy (Caserta) | 6 | 5 | 37 | 0.933 |
| Italy (Rome) | 19 | 5 | 40 | 0.649 |
| Spain+Portugal | 9 | 4 | 36 | 0.583 |
| New Zealand | 4 | 1 | 0 | 0.000 |
The assessment of the amino acid sequences revealed that all Xaj strains show a triplet deletion within the rpoD gene fragment, corresponding to valine, when compared with the corresponding rpoD sequence of X. axonopodis pv. citri strain 306. In addition, three Xaj strains all isolated in 2008 in Rome, Italy, from leaf lesions of three different walnut cultivars, namely CRA-FRU 66, 68 and 85 (i.e., phylotype 4), show an additional triplet deletion corresponding to arginine.
Figure 3.
Radial dendrogram using concatenated data and the Neighbor-joining (NJ) algorithm obtained with acnB, gapA, gyrB and rpoD nucleotidic gene sequences. Bootstrap values are reported at the main nodes. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
Figure 3.
Radial dendrogram using concatenated data and the Neighbor-joining (NJ) algorithm obtained with acnB, gapA, gyrB and rpoD nucleotidic gene sequences. Bootstrap values are reported at the main nodes. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
It is noteworthy that the dendrogram built with amino acid sequences identified the same four phylotypes found using the ML algorithm (
Figure 4). For representative strains of each phylotype (
i.e., NCPPB 411, NCPPB 1659, DAR 76583, CRA-FRU 33, CRA-FRU 61, CRA-FRU 66, CRA-FRU 75, CRA-FRU 80, CRA-FRU 87, NA 0823, WR 252b), the sequences of the four gene fragments were deposited at the NCBI databank. Their accession numbers are the following:
acnB: from FN667744 to FN667754;
gapA from FN667755 to FN667765;
gyrB from FN667766 to FN667776;
rpoD from FN667777 to FN667787.
Figure 4.
Dendrogram of relationships among X. a. pv. juglandis strains obtained by using the amino acid sequences of acnB, gapA, gyrB and rpoD. Bootstrap values are reported at the main nodes. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
Figure 4.
Dendrogram of relationships among X. a. pv. juglandis strains obtained by using the amino acid sequences of acnB, gapA, gyrB and rpoD. Bootstrap values are reported at the main nodes. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
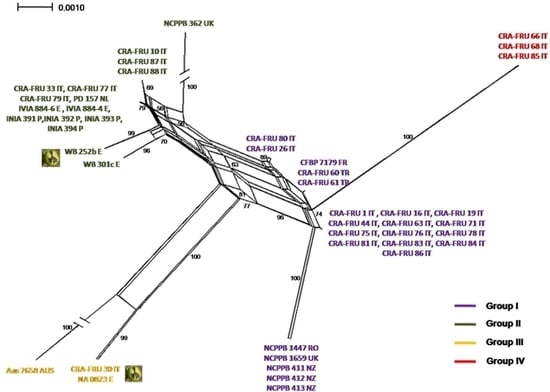
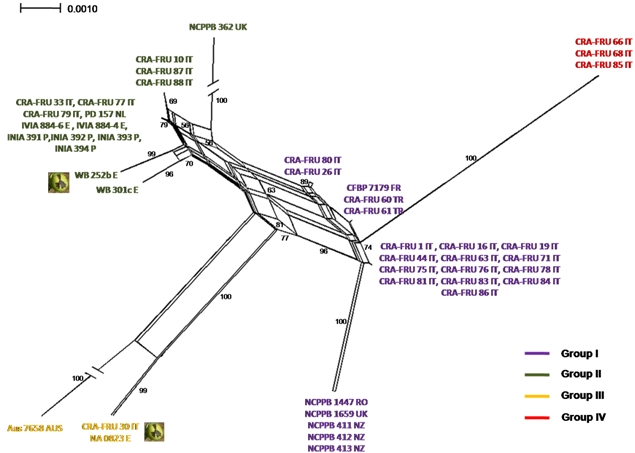
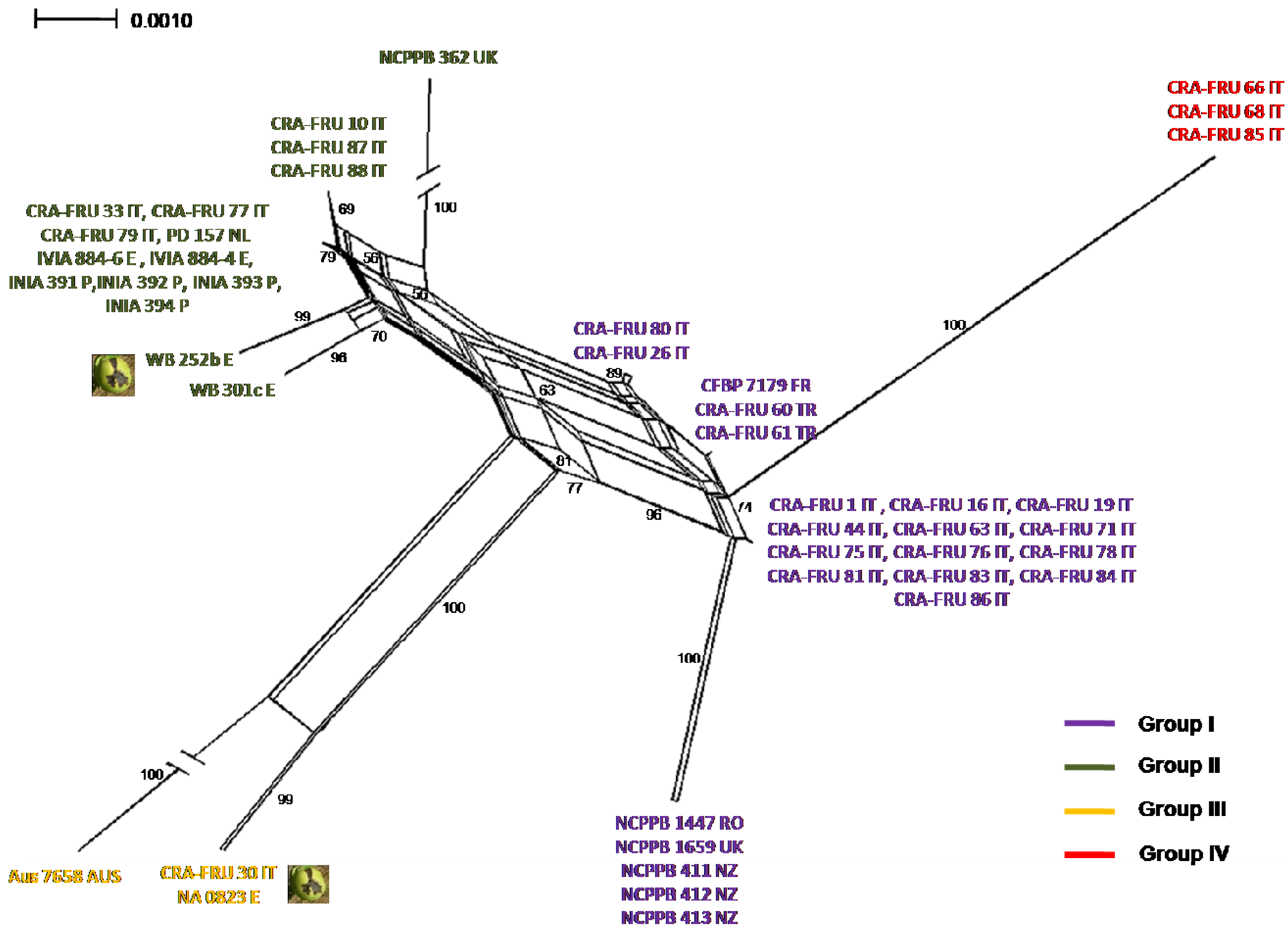
Phylogenetic networks were constructed using the Neighbor-Net algorithm to highlight conflicting signals in the gene sequence data, which would suggest the exchange or acquisition of genetic material among
Xaj strains. In a phylogenetic network, alternative phylogenies are represented by splits or reticulation. By definition, the more reticulation there is in a network, the more conflicting signals exist in the sample, possibly due to exchange of genetic material. The phylogenetic network obtained using the MLST concatenated data is shown in
Figure 5. A relevant reticulation was found connecting strains of all phylotypes. This result suggests that possible recombination events can occur within
Xaj populations.
Figure 5.
NeighborNet analysis of the concatenated set of four housekeeping genes, acnB, gapA, gyrB and rpoD of X. a. pv. juglandis strains. Bootstrap confidence values are shown along each branch. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
Figure 5.
NeighborNet analysis of the concatenated set of four housekeeping genes, acnB, gapA, gyrB and rpoD of X. a. pv. juglandis strains. Bootstrap confidence values are shown along each branch. The X. a. pv. juglandis strains isolated from apical necrosis of walnut fruits are pointed out.
To further investigate possible recombination events in
Xaj strains, we subsequently applied the GARD program, a likelihood-based model selection procedure detecting recombination breakpoints in the gene fragments. A breakpoint was predicted within
rpoD based on the AIC. This finding suggests that the reticulation pointed out with the NeighborNet analysis can be explained with the occurrence recombination events within
Xaj strains.
Table 4 shows the GARD output for all genes.
Table 4.
Recombination breakpoints predicted in analyzed gene fragment using GARD (Genetic Algorithm for Recombination Detection) and the Akaike Information Criterion (AIC).
Table 4.
Recombination breakpoints predicted in analyzed gene fragment using GARD (Genetic Algorithm for Recombination Detection) and the Akaike Information Criterion (AIC).
| Gene | Number of breakpoints | Lenght of fragment for each partition (nt) | NeighborNet results for partition | Number of detectable breakpoints/Total of breakpoints (P < 0.05) ** |
|---|
| acnB | 0 | NA | NA | NA |
| gapA | 0 | NA | NA | NA |
| gyrB | 0 | NA | NA | NA |
| rpoD | 1 * | 156* | reticulation | 1/1 |
To test the possible role of homologous recombination in Xaj strains, we checked the level of linkage disequilibrium in our sample. If the loci are under linkage equilibrium, then high rates of recombination are operating in the population. The standardized index of association ISA measures the extent of the linkage, and it is equal or very close to zero when the strains are experiencing free recombination. By contrast, if this value is significantly different from zero, the population is considered clonal (i.e., linkage disequilibrium). The assessment of linkage disequilibrium at the pathovar level indicated that the Xaj strains tested here are clonal because the ISA value, although relatively low (0.32) was statistically different from 0 (i.e., VD: 1.836 > LMC: 1.047).
3.2. Selection Analysis and Gene Flow
The results of the McDonald-Kreitman test are shown in
Table 5. This analysis revealed that in
acnB and
gapA gene fragments of
Xaj strains there is a very large amount of non-neutral polymorphism. This data confirm that a possible different selection is acting in the
Xaj strains isolated from different walnut organs. With the aim of investigating the evolution model that best fit our sample, we also performed the Tajima’s D and the Fu and Li’s D tests and evaluated their statistical significance. Both tests indicated that all four genes and the
Xaj strains isolated in Italy, Spain and Portugal are currently under neutral selection. In fact, in each case, the negative values obtained are not statistically significant. By contrast, the four
Xaj strains obtained from fruit apical necrosis, showed high significant positive values for both the Tajima’s and Fu and Li’s D tests, indicating diversifying selection (
Table 6).
Table 5.
Neutral and non-neutral polymorphism between Xanthomonas arboricola pv. juglandis strains isolated from walnut “apical necrosis” of fruits and the other strains according to the Mc Donald-Kreitman test. The test provides the ratio between non-synonymous amino acids and synonymous amino acids.
Table 5.
Neutral and non-neutral polymorphism between Xanthomonas arboricola pv. juglandis strains isolated from walnut “apical necrosis” of fruits and the other strains according to the Mc Donald-Kreitman test. The test provides the ratio between non-synonymous amino acids and synonymous amino acids.
| | Neutral | Non-neutral | Total |
|---|
| acn B | 2 | 52 | 54 |
| gap A | 0 | 19 | 19 |
| gyt B | 18 | 0 | 18 |
| rpo D | 34 | 2 | 36 |
We also investigated the CAI. High CAI values correlate with high levels of gene expression, and average or low CAI values correlate with low levels of gene expression. The CAI value for all of the four genes was high, varying from 0.805 to 0.855, and it was statistically significant, as shown by the corresponding E-CAI values always being lower than the CAI values (
Table 6). The gene flow among
Xaj strains isolated from different geographic areas was estimated using the F
ST test (
Table 7). This value indicated that the genetic differentiation between strains from different areas is rather low. This does not support the hypothesis of geographic isolation, even though the genetic differentiation between populations increases as the geographic distance between them increases with the strains from New Zealand showing the highest differentiation (
Table 7).
Table 6.
Selection tests, Codon Adaptation Index (CAI) and expected CAI (E-CAI) observed in four Xanthomonas arboricola pv. juglandis housekeeping gene fragments at gene and at strain geographic origin levels. AN: X. a. pv. juglandis strains obtained from walnut fruits showing “apical necrosis” symptoms.
Table 6.
Selection tests, Codon Adaptation Index (CAI) and expected CAI (E-CAI) observed in four Xanthomonas arboricola pv. juglandis housekeeping gene fragments at gene and at strain geographic origin levels. AN: X. a. pv. juglandis strains obtained from walnut fruits showing “apical necrosis” symptoms.
| Gene/Origin of strains | Tajima’s D | Fu and Li’s D | CAI | E-CAI |
|---|
| acnB | −0.3630* | −0.2351* | 0.805 | 0.743 |
| gapA | −1.3869* | −3.9380* | 0.819 | 0.787 |
| gyrB | −1.2075* | −1.4734* | 0.855 | 0.789 |
| rpoD | −1.1436* | −0.0671* | 0.823 | 0.745 |
| Italy | −0.3346* | −0.1122* | NA | NA |
| Spain+Portugal | −1.6609* | −1.8527* | NA | NA |
| AN strains | 1.5571** | 1.6086** | NA | NA |
Table 7.
Gene flow estimates (FST) among Xanthomona arboricola pv. juglandis strains isolated from different geographic areas.
Table 7.
Gene flow estimates (FST) among Xanthomona arboricola pv. juglandis strains isolated from different geographic areas.
| | FST |
|---|
| New Zealand vs. Italy | 0.47561 |
| New Zealand vs. Spain + Portugal | 0.61021 |
| Italy vs Spain + Portugal | 0.34149 |
| Italy (Rome) vs. Italy (Caserta) | 0.26806 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


