Understanding the Extent and Sources of Variation in Gut Microbiota Studies; a Prerequisite for Establishing Associations with Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Gut Microbiota and GI Diseases
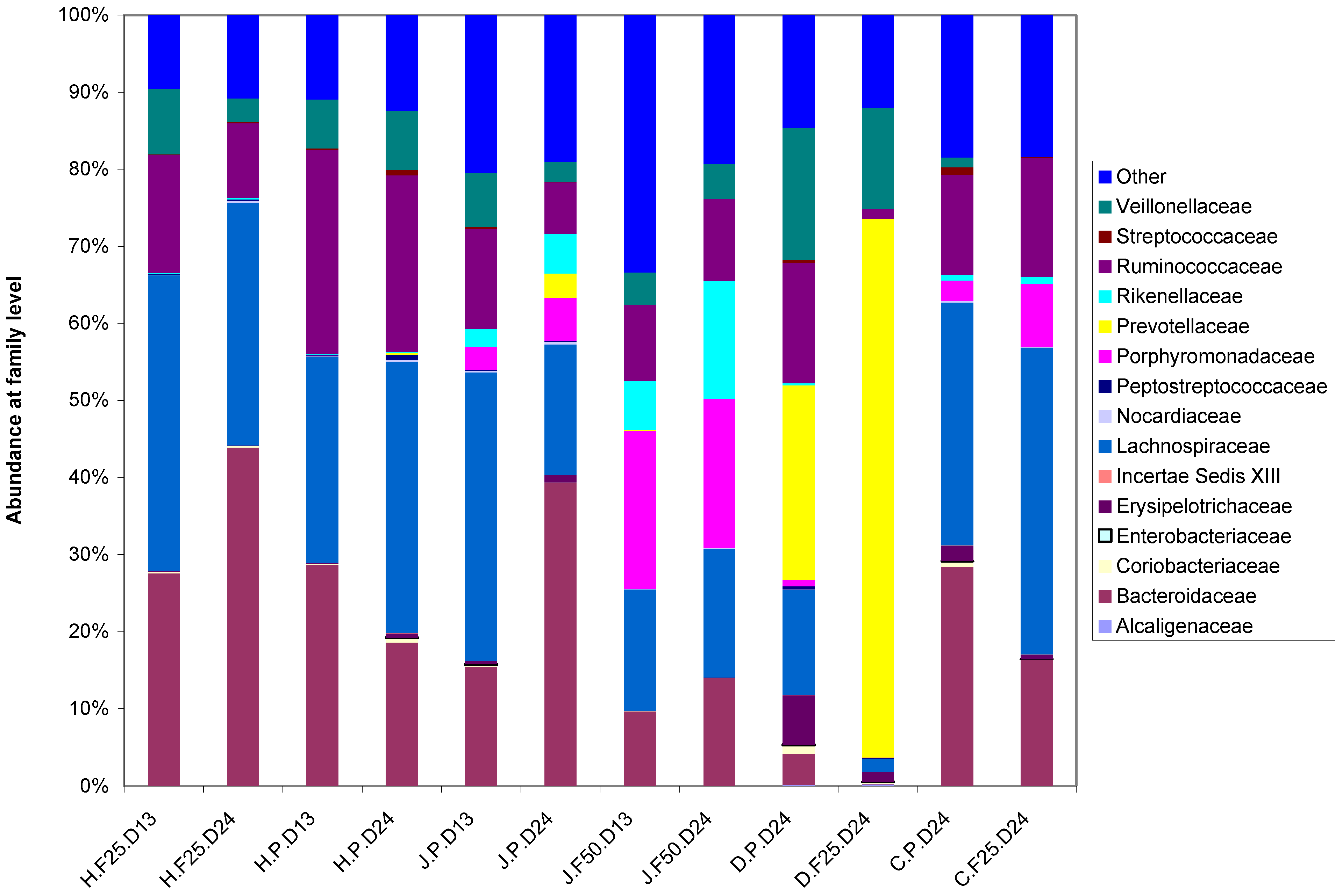
1.2. Gut microbiota Variation and Dynamics
1.3. The Core Microbiome
2. Barriers in Population Based Microbiota Studies
2.1.Gut Microbiota Sampling

2.2. Understanding Microbiota Dynamics in Studies with Controlled Diets


2.3.The Need for Standardization of Analysis Methodology
3. Conclusions
Acknowledgements
References and notes
- Metchnikoff, E. The Prolongation of Life; Heinemann: London, UK, 1907; pp. 73–84. [Google Scholar]
- Dethlefsen, L.; Eckburg, P.B.; Bik, E.M.; Relman, D.A. Assembly of the human intestinal microbiota. Trends Ecol. Evol. 2006, 21, 517–523. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar]
- Lunn, J.C.; Kuhnle, G.; Mai, V.; Frankenfeld, C.; Shuker, D.E.; Glen, R.C.; Goodman, J.M.; Pollock, J.R.; Bingham, S.A. The effect of haem in red and processed meat on the endogenous formation of N-nitroso compounds in the upper gastrointestinal tract. Carcinogenesis 2007, 28, 685–690. [Google Scholar]
- Khachatryan, Z.A.; Ktsoyan, Z.A.; Manukyan, G.P.; Kelly, D.; Ghazaryan, K.A.; Aminov, R.I. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE 2008, 3, e3064. [Google Scholar]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Jewell, D.; Satsangi, J.; Mathew, C.G.; Parkes, M.; Georges, M.; Daly, M.J. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host. Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Dore, J. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm. Bowel. Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Vaneechoutte, M.; Doerffel, Y. Active Crohn's disease and ulcerative colitis can be specifically diagnosed and monitored based on the biostructure of the fecal flora. Inflamm. Bowel. Dis. 2008, 14, 147–161. [Google Scholar] [CrossRef]
- Mai, V.; Colbert, L.H.; Perkins, S.N.; Schatzkin, A.; Hursting, S.D. Intestinal microbiota: a potential diet-responsive prevention target in ApcMin mice. Mol. Carcinog. 2007, 46, 42–48. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; Clune, Y.; Collins, J.K.; O'Sullivan, G.C.; O'Riordan, M.; Holmes, E.; Wang, Y.; Marchesi, J.R. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ. Microbiol. 2008, 10, 789–798. [Google Scholar] [CrossRef]
- Mai, V.; Draganov, P.V. Recent advances and remaining gaps in our knowledge of associations between gut microbiota and human health. World J. Gastroenterol. 2009, 15, 81–85. [Google Scholar] [CrossRef]
- Culpepper, T.; Vedam-Mai, V.; Valentine, J.; Polyak, S.; Mai, V. Gut Microbiota composition correlates with Colorectal Polyp Prevalence. In Proceedings of the 110th General Meeting American Society for Microbiology, 23-27 Mai 2010; ASM: San Diego, CA, USA.
- Young, C.; Sharma, R.; Handfield, M.; Mai, V.; Neu, J. Biomarkers for infants at risk for necrotizing enterocolitis: clues to prevention? Pediatr. Res. 2009, 65, 91R–97R. [Google Scholar] [CrossRef]
- Mshvildadze, M.; Neu, J.; Shuster, J.; Theriaque, D.; Li, N.; Mai, V. Intestinal microbial ecology in premature infants assessed with non-culture-based techniques. J. Pediatr. 2009, 156, 20–25. [Google Scholar]
- Neu, J.; Mshvildadze, M.; Mai, V. A roadmap for understanding and preventing necrotizing enterocolitis. Curr. Gastroenterol. Rep. 2008, 10, 450–457. [Google Scholar] [CrossRef]
- Favier, C.F.; De Vos, W.M.; Akkermans, A.D.L. Development of bacterial and bifidobacterial communities in feces of newborn babies. Anaerobe 2003, 9, 219–229. [Google Scholar] [CrossRef]
- Tannock, G.W. Molecular methods for exploring the intestinal ecosystem. Br. J. Nutr. 2002, 87, S199–S201. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Vaughan, E.E.; De Vos, W.M. A microbial world within us. Mol. Microbiol. 2006, 59, 1639–1650. [Google Scholar] [CrossRef]
- Frank, D.N.; Pace, N.R. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 2008, 24, 4–10. [Google Scholar] [CrossRef]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006, 7, 371. [Google Scholar] [CrossRef]
- Hamady, M.; Lozupone, C.; Knight, R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; Egholm, M.; Henrissat, B.; Heath, A.C.; Knight, R.; Gordon, J.I. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Sun, Y.; Cai, Y.; Liu, L.; Yu, F.; Farrell, M.L.; McKendree, W.; Farmerie, W. ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009, 37, e76. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Parkhill, J.; Weissenbach, J.; Bork, P.; Ehrlich, S.D.; Wang, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.P.; Wang, Y.; Sprenger, N.; Yap, I.K.; Lundstedt, T.; Lek, P.; Rezzi, S.; Ramadan, Z.; van Bladeren, P.; Fay, L.B.; Kochhar, S.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol. Syst. Biol. 2008, 4, 157. [Google Scholar]
- Martin, F.P.; Sprenger, N.; Yap, I.K.; Wang, Y.; Bibiloni, R.; Rochat, F.; Rezzi, S.; Cherbut, C.; Kochhar, S.; Lindon, J.C.; Holmes, E.; Nicholson, J.K. Panorganismal gut microbiome-host metabolic crosstalk. J. Proteome. Res. 2009, 8, 2090–2105. [Google Scholar] [CrossRef]
- Blaut, M.; Collins, M.D.; Welling, G.W.; Dore, J.; van Loo, J.; de Vos, W. Molecular biological methods for studying the gut microbiota: the EU human gut flora project. Br. J. Nutr. 2002, 87, S203–S211. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; von Wright, A.; Vilpponen-Salmela, T.; Ben Amor, K.; Akkermans, A.D.; de Vos, W.M. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ. Microbiol. 2002, 68, 3401–3407. [Google Scholar] [CrossRef]
- Croucher, L.J.; Bury, J.P.; Williams, E.A.; Riley, S.A.; Corfe, B.M. Commonly used bowel preparations have significant and different effects upon cell proliferation in the colon: a pilot study. BMC Gastroenterol. 2008, 8, 54. [Google Scholar] [CrossRef]
- Bucher, P.; Gervaz, P.; Egger, J.F.; Soravia, C.; Morel, P. Morphologic alterations associated with mechanical bowel preparation before elective colorectal surgery: a randomized trial. Dis. Colon. Rectum 2006, 49, 109–112. [Google Scholar]
- Swidsinski, A.; Loening-Baucke, V.; Verstraelen, H.; Osowska, S.; Doerffel, Y. Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 2008, 135, 568–579. [Google Scholar] [CrossRef]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjoberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; Stoel, M.; Zhou, Y.; Sodergren, E.; Weinstock, G.M.; Bevins, C.L.; Williams, C.B.; Bos, N.A. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2009, 11, 76–82. [Google Scholar]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med 2009, 1, 6ra14. [Google Scholar]
- Mai, V.; Katki, H.; Clevidence, B.; Hursting, S.; Harmsen, H.; Schatzkin, A. Changes in the fecal flora composition of human volunteers in a double-blind randomized black tea feeding study. J. Nutr. 2002, 132, 3551S. [Google Scholar]
- Salonen, A.; Nikkila, J.; Jalanka-Tuovinen, J.; Immonen, O.; Rajilic-Stojanovic, M.; Kekkonen, R.A.; Palva, A.; De Vos, W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 2010, 81, 127–134. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mai, V.; Ukhanova, M.; Baer, D.J. Understanding the Extent and Sources of Variation in Gut Microbiota Studies; a Prerequisite for Establishing Associations with Disease. Diversity 2010, 2, 1085-1096. https://doi.org/10.3390/d2091085
Mai V, Ukhanova M, Baer DJ. Understanding the Extent and Sources of Variation in Gut Microbiota Studies; a Prerequisite for Establishing Associations with Disease. Diversity. 2010; 2(9):1085-1096. https://doi.org/10.3390/d2091085
Chicago/Turabian StyleMai, Volker, Maria Ukhanova, and David J. Baer. 2010. "Understanding the Extent and Sources of Variation in Gut Microbiota Studies; a Prerequisite for Establishing Associations with Disease" Diversity 2, no. 9: 1085-1096. https://doi.org/10.3390/d2091085