
Potential Population Genetic Consequences of Habitat Fragmentation in Central European Forest Trees and Associated Understorey Species—An Introductory Survey


Abstract
:1. Introduction
1.1. The Foundation: Basics of Population Genetics
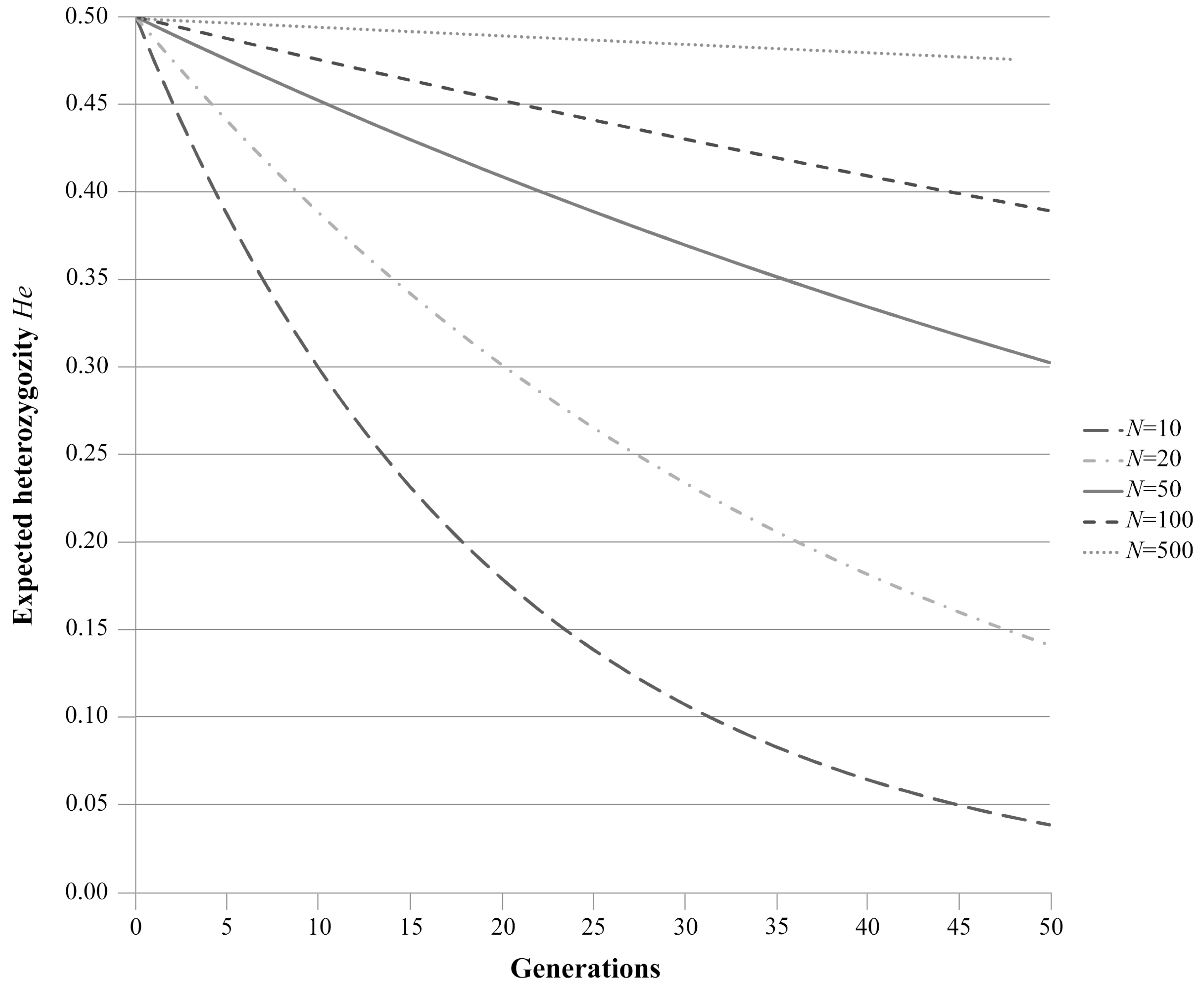
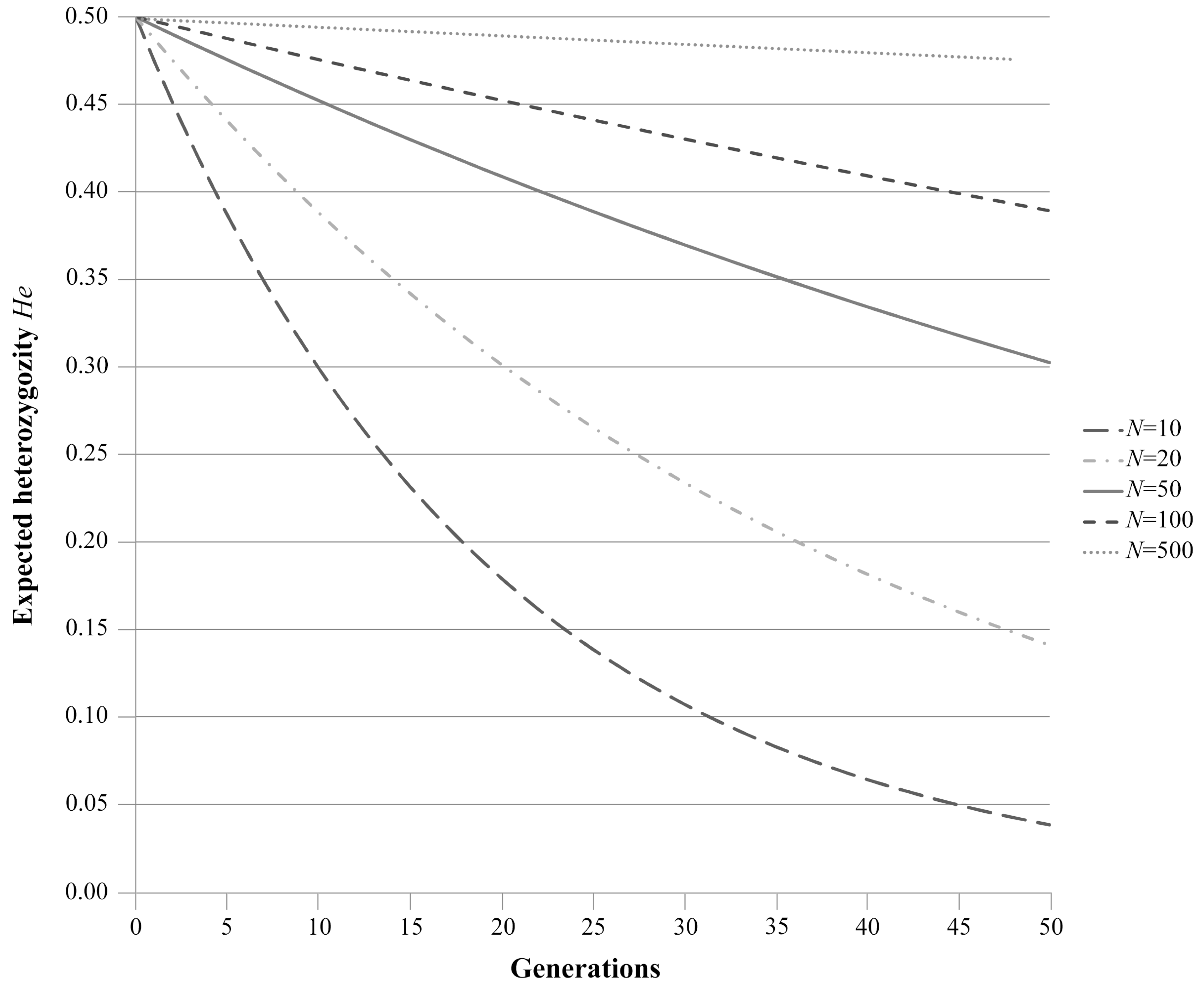
1.2. Random Genetic Drift
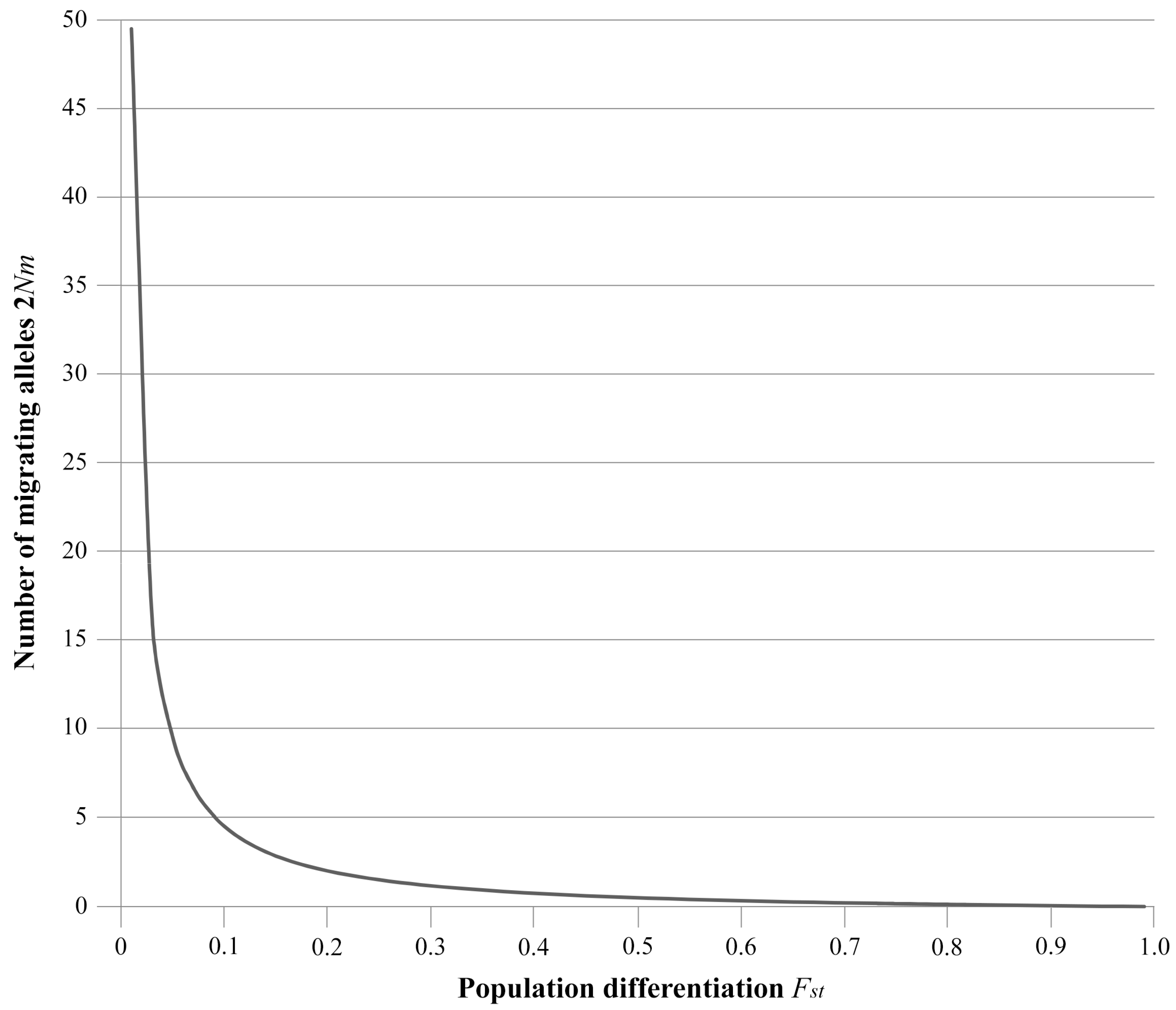
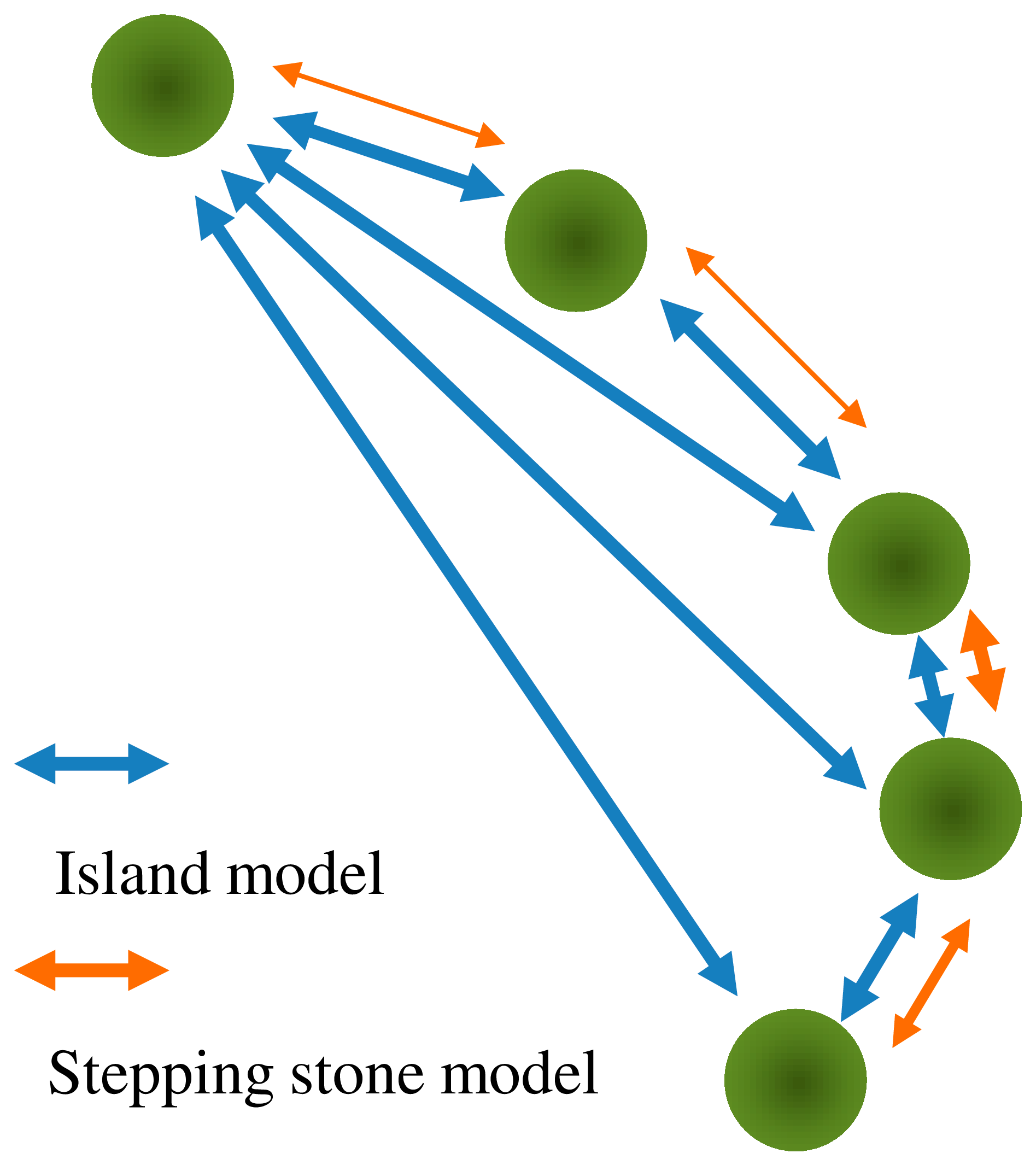
1.3. Gene Flow
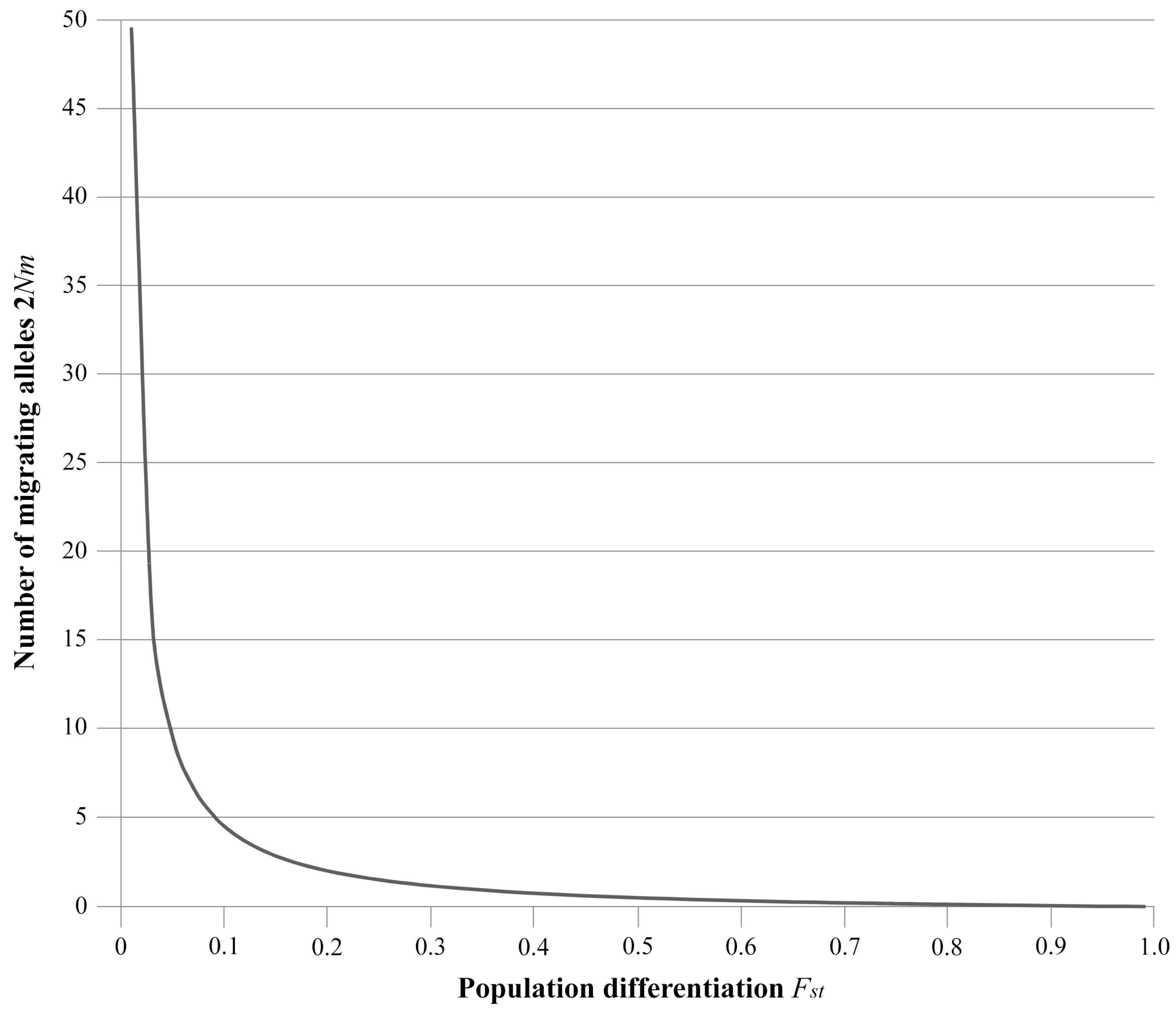
1.4. Desirable Levels of Gene Flow
1.5. Alternative Approaches: Estimating Inbreeding and Allee Effects
2. Population Genetic Characteristics of Central European Forest Tree Species
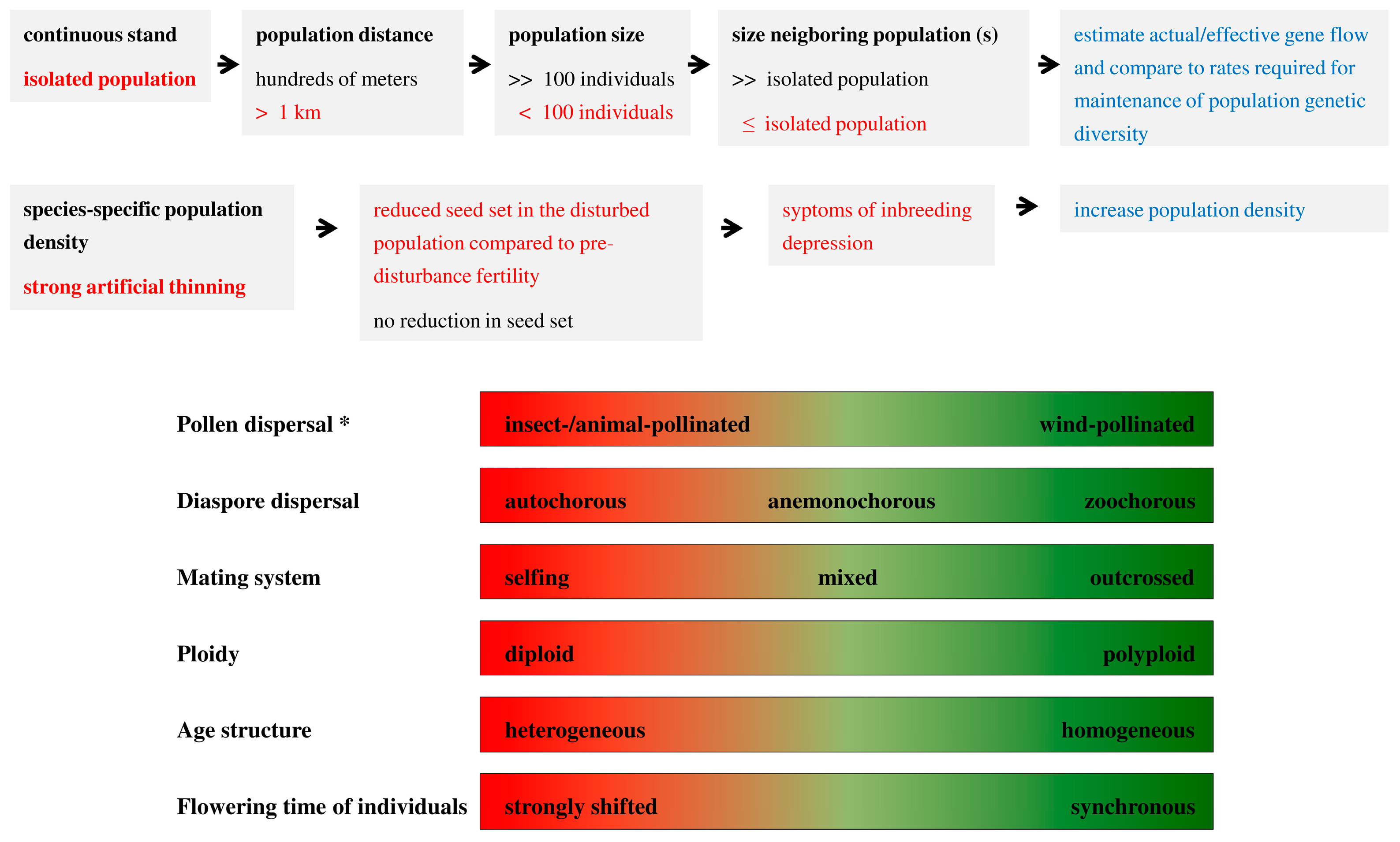
2.1. The Relative Importance of Seeds and Pollen for Gene Flow
2.2. Modes of Seed and Pollen Dispersal and Its Significance for Gene Flow
2.3. Mating and Inbreeding
2.4. Population Fragmentation in Forest Understorey Plant Species
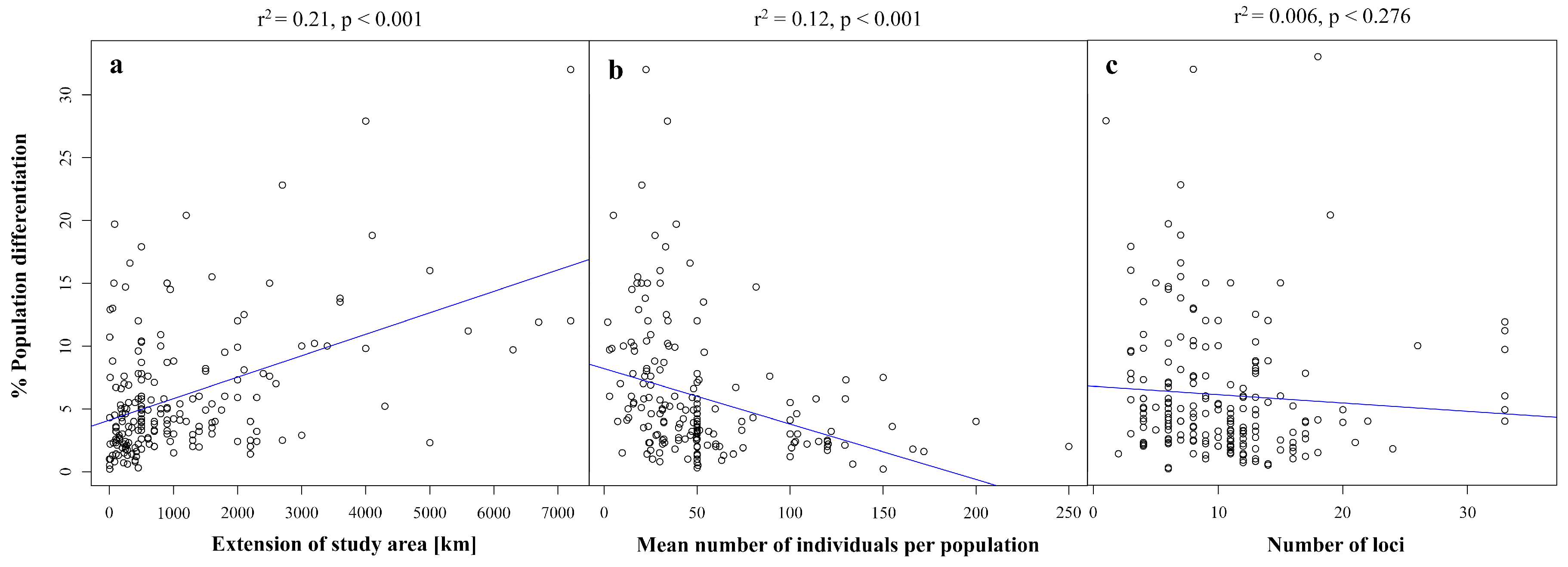
3. Effects of Study Design on Population Genetic Differentiation
4. Conclusions and Recommendations
4.1. Spatial Distance among and Arrangement of Populations Reduce Gene Flow
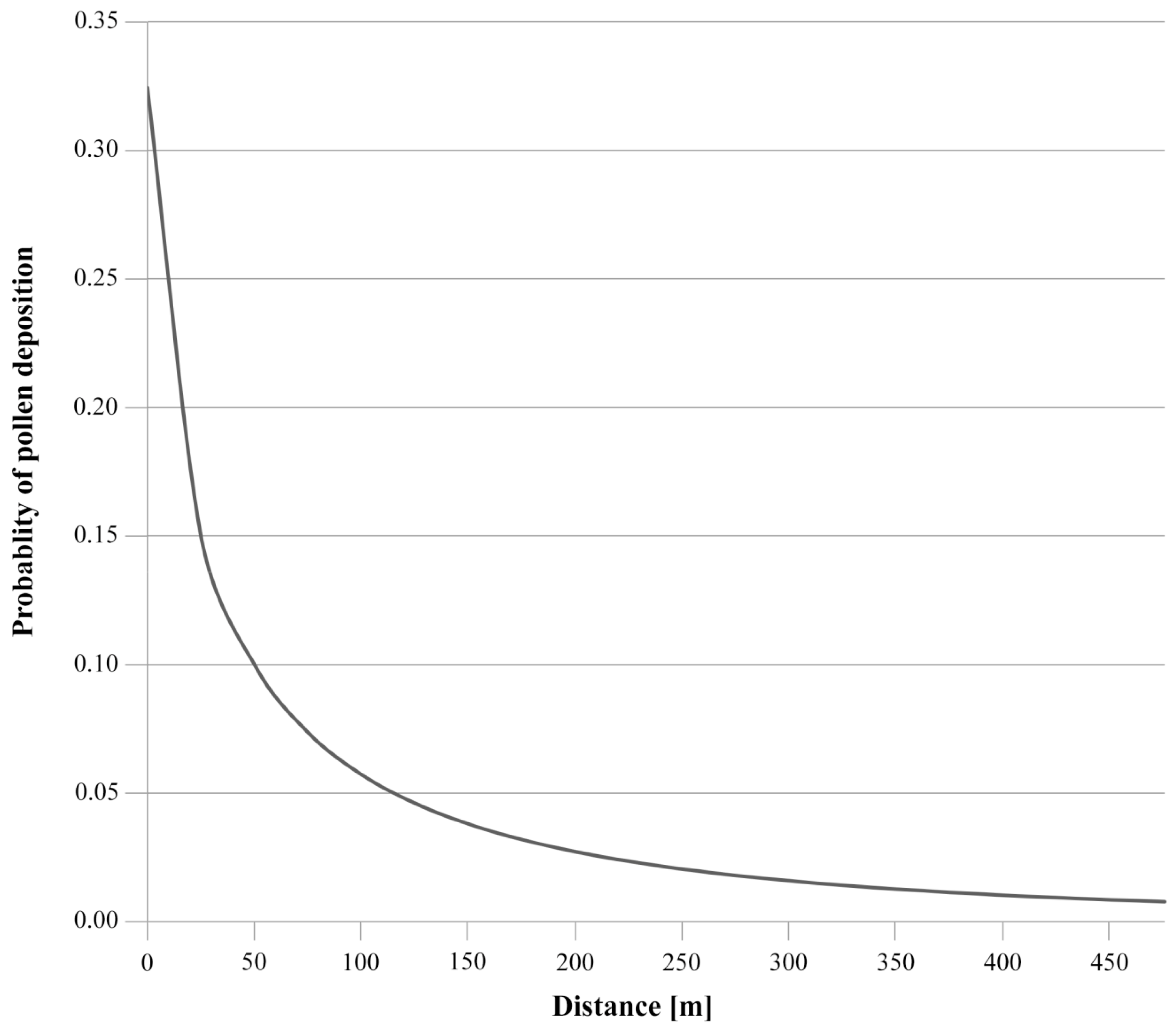
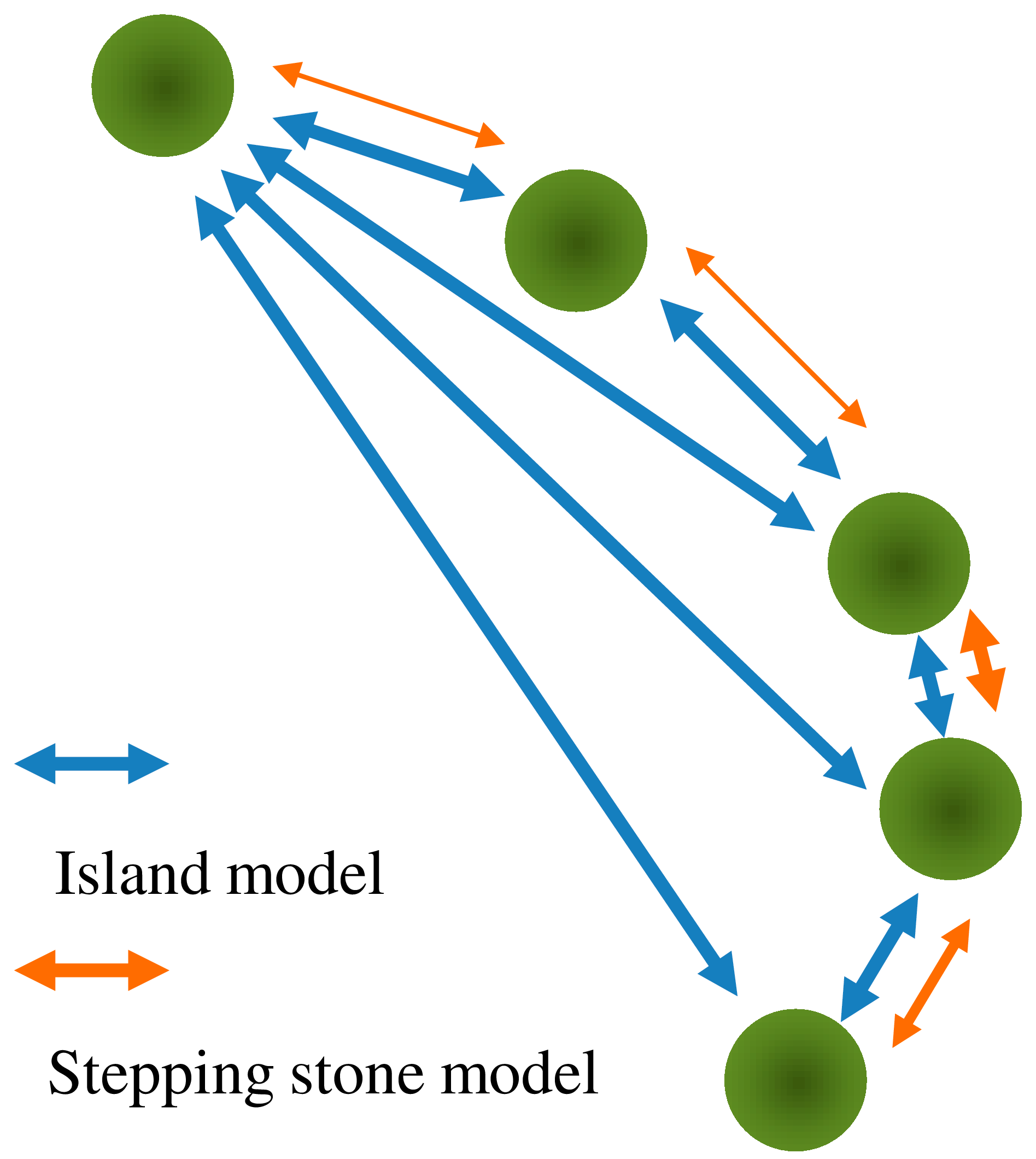
4.2. Superimposition of Pollen Increases Gene Flow
4.3. Gene Flow versus Random Genetic Drift
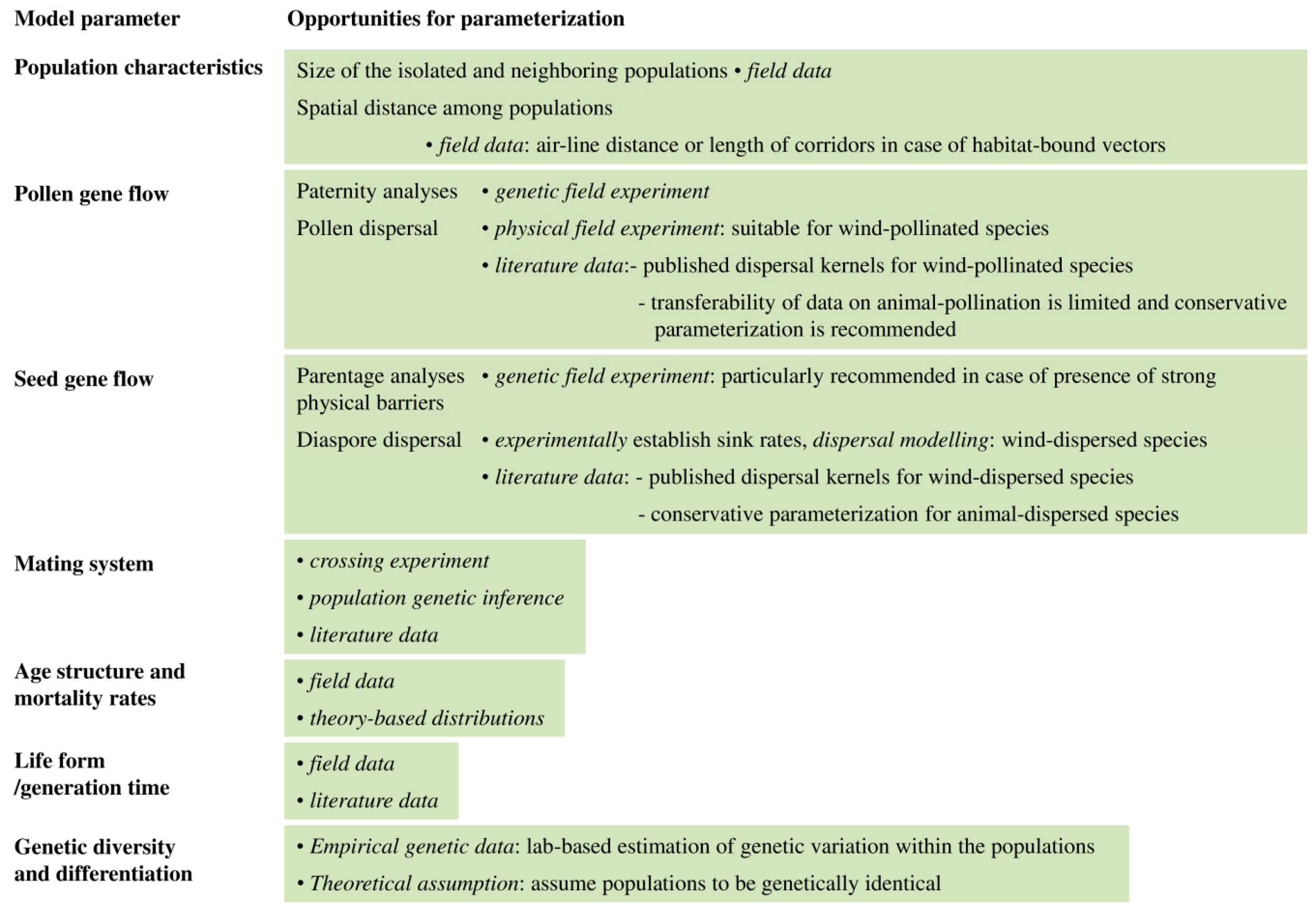
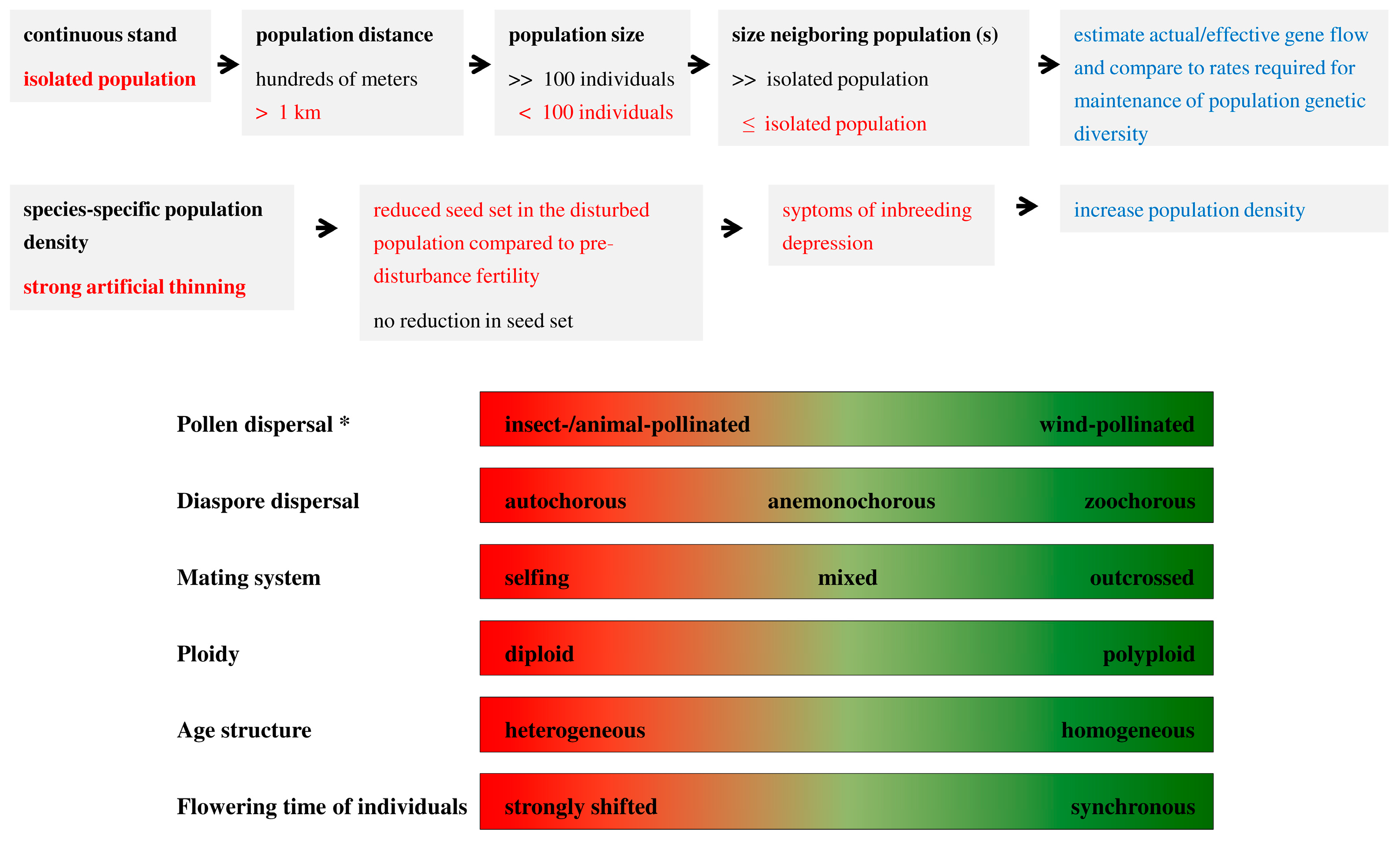
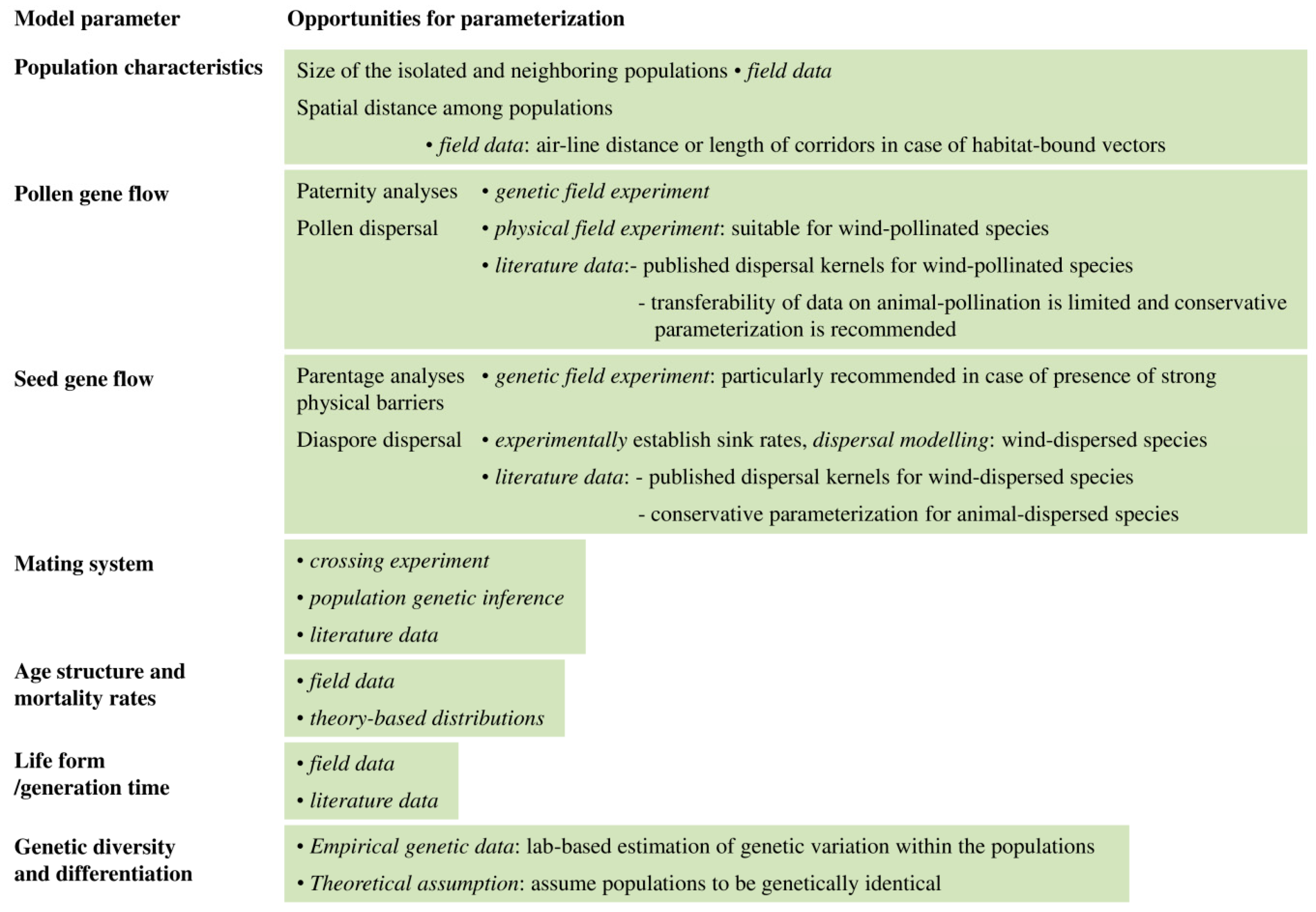
4.4. Recommendations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williams, M. Deforesting the Earth. From Prehistory to Global Crisis; University of Chicago Press: Chicago, IL, USA, 2003; p. 689. [Google Scholar]
- Mather, A. The transition from deforestation to reforestation in Europe. In Agricultural Technologies and Tropical Deforestation; Angelsen, A., Kaimowitz, D., Eds.; Center for International Forestry Research (CIFOR): Bogor, Indonesia, 2001; pp. 35–52. [Google Scholar]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Jump, A.; Peñuelas, P. Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc. Natl. Acad. Sci. USA 2006, 103, 8096–8100. [Google Scholar] [CrossRef] [PubMed]
- Vakkari, P.; Rusanen, M.; Raisio, J.; Toivonen, H. Genetic variability of fragmented stands of pedunculate oak (Quercus robur) in Finland. Genetica 2006, 127, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Bacles, C.F.E.; Burczyk, J.; Lowe, A.J. Historical and contemporary mating patterns in remnant populations of the forest tree Fraxinus excelsior L. Evolution 2005, 59, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.T.; Ison, J.L.; Ashley, M.V.; Howe, H.F. The paradox of forest fragmentation genetics. Conserv. Biol. 2008, 22, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. B 1996, 351, 1291–1298. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 124. [Google Scholar] [CrossRef]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Ehrendorfer, F. (Ed.) Liste der Gefäßpflanzen Mitteleuropas, 2nd ed.; Gustav Fischer: Stuttgart, Germany, 1973; p. 318.
- Johannsen, W. Über die Erblichkeit in Populationen und in reinen Linien. In Ein Beitrag zur Beleuchtung Schwebender Selektionsfragen; Fischer-Verlag: Jena, Germany, 1903; p. 76. [Google Scholar]
- Schlötterer, Ch. Microsatellites. In Molecular Genetic Analysis of Populations, 2nd ed.; Hoelzel, A.R., Ed.; Oxford University Press: New York, NY, USA, 1998; pp. 237–261. [Google Scholar]
- Rothe, G.M. Electrophoresis of enzymes. Laboratory Methods; Springer: Berlin, Germany, 1994; p. 307. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; p. 512. [Google Scholar]
- Gregorius, H.-R. The concept of genetic diversity and its formal relationship to heterozygosity and genetic distance. Math. Biosci. 1978, 41, 253–271. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114–138. [Google Scholar] [PubMed]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Speciation, hybrid zones and phylogeography—Or seeing genes in space and time. Mol. Ecol. 2001, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 3rd ed.; Sinauer: Sunderland, MA, USA, 1997; p. 542. [Google Scholar]
- Wang, J. Application of the One-Migrant-per-Generation Rule to conservation and management. Conserv. Biol. 2004, 18, 332–343. [Google Scholar] [CrossRef]
- Loveless, M.D.; Hamrick, J.L. Ecological determinants of genetic structure in plant populations. Ann. Rev. Ecol. Syst. 1984, 15, 65–95. [Google Scholar] [CrossRef]
- Avise, J.C. Molecular Markers, Natural History and Evolution, 2nd ed.; Sinauer: Sunderland, MA, USA, 2004; p. 684. [Google Scholar]
- Petit, R.J.; Hampe, A. Some evolutionary consequences of being a tree. Ann. Rev. Ecol. Evol. Syst. 2006, 37, 187–214. [Google Scholar] [CrossRef]
- Ellstrand, N.C. Gene flow among seed plant populations. New For. 1992, 6, 241–256. [Google Scholar] [CrossRef]
- Katul, G.G.; Porporato, A.; Nathan, R.; Siqueira, M.; Soons, M.B.; Poggi, D.; Horn, H.S.; Levins, S.A. Mechanistic analytical models for long-distance seed dispersal by wind. Am. Nat. 2005, 166, 368–381. [Google Scholar] [PubMed]
- Straka, H. Pollen- und Sporenkunde; Gustav Fischer: Sunderland, MA, USA, 1975; p. 238. [Google Scholar]
- Eisenhut, G. Untersuchungen über die Morphologie und Ökologie der Pollenkörner heimischer und fremdländischer Waldbäume. Forstwiss. For. 1961, 15, 1–68. [Google Scholar]
- Handel, S.N. Pollination Ecology, Plant Population Structure, and Gene Flow. Pollination Biology; Real, L., Ed.; Academic Press: Orlando, FL, USA, 1983; pp. 163–211. [Google Scholar]
- Goto, S.; Shimatani, K.; Yoshimaru, H.; Takahashi, Y. Fat-tailed gene flow in the dioecious canopy tree species Fraxinus mandshurica var. japonica revealed by microsatellites. Mol. Ecol. 2006, 15, 2985–2996. [Google Scholar] [PubMed]
- Streiff, R.; Ducousso, A.; Lexer, Ch.; Steinkellner, H.; Glössl, J.; Kremer, A. Pollen dispersal inferred from paternity analysis in a mixed oak stand of Quercus robur L. and Q. petraea (Matt.) Liebl. Mol. Ecol. 1999, 8, 831–841. [Google Scholar] [CrossRef]
- Adams, W.T.; Griffin, A.R.; Moran, G.F. Using paternity analysis to measure effective pollen dispersal in plant populations. Am. Nat. 1992, 140, 762–780. [Google Scholar] [CrossRef] [PubMed]
- Burczyk, J.; Adams, W.T.; Moran, G.F.; Griffin, A.R. Complex patterns of mating revealed in a Eucalyptus regnans seed orchard using allozyme markers and the neighborhood model. Mol. Ecol. 2002, 11, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Smouse, P.E.; Meagher, T.R.; Kobak, C.J. Parentage analysis in Chamaelirium luteum (L.): Why do some males have disproportionate reproductive contributions? J. Evol. Biol. 1999, 12, 1069–1077. [Google Scholar] [CrossRef]
- Austerlitz, F.; Dick, C.W.; Dutech, C.; Klein, E.K.; Oddou-Muratorio, S.; Smouse, P.E.; Sork, V.L. Using genetic markers to estimate the pollen dispersal curve. Mol. Ecol. 2004, 13, 937–954. [Google Scholar] [CrossRef] [PubMed]
- Grivet, D.; Robledo-Arnuncio, J.J.; Smouse, P.E.; Sork, V.L. Relative contribution of contemporary pollen and seed dispersal to the effective parental size of seedling population of California valley oak (Quercus lobata Née). Mol. Ecol. 2009, 18, 3967–3979. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Chadoeuf, J.; Gugerli, F.; Lascoux, M.; Buiteveld, J.; Cottrell, J.; Dounavi, A.; Fineschi, S.; Forrest, L.L.; Fogelqvist, J.; et al. High rates of gene flow by pollen and seed in oak populations across Europe. PLoS ONE 2014, 9, e85130. [Google Scholar] [CrossRef]
- Robledo-Arnuncio, J.J.; Klein, E.K.; Muller-Landau, H.C.; Santamaria, L. Space, time and complexity in plant dispersal ecology. Mov. Ecol. 2014, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Burczyk, J.; Adams, W.T.; Birkes, D.S.; Chybicki, I.J. Using genetic markers to directly estimate gene flow and reproductive success parameters in plants on the basis of naturally regenerated seedlings. Genetics 2006, 173, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.V.; Clark, J.S. Estimating seed and pollen movement in a monoecious plant: A hierarchical Bayesian approach integrating genetic and ecological data. Mol. Ecol. 2011, 20, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Chybicki, I.J.; Burczyk, J. Seeing the forest through the trees: Comprehensive inference on individual mating patterns in a mixed stand of Quercus robur and Q. petraea. Ann. Bot. (Lond.) 2013, 112, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Robledo-Arnuncio, J.J. Joint estimation of contemporary seed and pollen dispersal rates among plant populations. Mol. Ecol. Res. 2012, 12, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Burczyk, J.; DiFazio, S.P.; Adams, W.T. Gene flow in forest trees: How far do genes really travel? For. Genet. 2004, 11, 179–192. [Google Scholar]
- Krutovsky, K.V.; Burczyk, J.; Chybicki, I.; Finkeldey, R.; Pyhäjärvi, T.; Robledo-Arnuncio, J.J. Gene Flow, Spatial Structure, Local Adaptation, and Assisted Migration in Trees. In Genomics of Tree Crops; Schnell, R.J., Priyadarshan, P.M., Eds.; Springer: New York, NY, USA, 2012; pp. 71–116. [Google Scholar]
- Wright, S. The evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [PubMed]
- Slatkin, M.; Barton, N.H. A comparison of three different methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef]
- Smouse, P.E.; Sork, V.L. Measuring pollen flow in forest trees: an exposition of alternative approaches. For. Ecol. Manag. 2004, 197, 21–38. [Google Scholar] [CrossRef]
- Sork, V.L.; Nason, J.; Campell, D.R.; Fernández, J.F. Landscape approaches to historical and contemporary gene flow in plants. Trends Ecol. Evol. 1999, 14, 219–224. [Google Scholar] [CrossRef]
- Hudson, R.R. Island models and the coalescent process. Mol. Ecol. 1998, 7, 413–418. [Google Scholar] [CrossRef]
- Govindaraju, D.R. Estimates of gene flow in forest trees. Biol. J. Linn. Soc. 1989, 37, 345–357. [Google Scholar] [CrossRef]
- Vekemans, X.; Hardy, O.J. New insights from fine-scale spatial genetic structure analyses in plant populations. Mol. Ecol. 2004, 13, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Hardy, O.; Maggia, L.; Bandou, E.; Breyne, P.; Caron, H.; Chevallier, M.-H.; Doligez, A.; Dutech, C.; Kremer, A.; Latouche-Hallé, C.; et al. Fine-scale genetic structure and gene dispersal inferences in 10 Neotropical tree species. Mol. Ecol. 2006, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [PubMed]
- Samarasin, P.; Shuter, B.J.; Wright, S.I.; Rodd, F.H. The problem of estimating recent genetic connectivity in a changing world. Conserv. Biol. 2016, 31, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Breeding structure of populations in relation to speciation. Am. Nat. 1940, 74, 232–248. [Google Scholar] [CrossRef]
- Scott Mills, L.; Allendorf, F.W. The one-migrant-per generation rule in conservation and managment. Conserv. Biol. 1996, 10, 1509–1518. [Google Scholar] [CrossRef]
- Charlesworth, D.; Charlesworth, B. Inbreeding depression and its evolutionary consequences. Ann. Rev. Ecol. Syst. 1987, 18, 237–268. [Google Scholar] [CrossRef]
- Richards, A.J. Plant Breeding Systems, 2nd ed.; Chapman & Hall: London, UK, 1997; p. 529. [Google Scholar]
- Young, A.; Boyle, T.; Brown, T. The population genetic consequences of habitat fragmentation for plants. Trends Ecol. Evol. 1996, 11, 413–418. [Google Scholar] [CrossRef]
- Oostermeijer, J.G.B.; Berholz, A.; Poschlod, P. Genetical aspects of fragmented populations. In Species Survival in Fragmented Landscapes; Settele, J.C.R., Margules, P., Poschlod, P., Henle, K., Eds.; Kluwer: Dordrecht, The Netherlands, 1996; pp. 93–101. [Google Scholar]
- De Nettancourt, D. Incompatibility and Incongruity in Wild and Cultivated Plants, 2nd ed.; Springer: Berlin, Germany, 2001; p. 322. [Google Scholar]
- Kamm, U.; Gugerli, F.; Rotach, P.; Edwards, P.; Holderegger, R. Seltenes und zerstreutes Vorkommen: Auswirkungen auf den Paarungserfolg des Speierlings. Schweiz. Z. Forstwes. 2012, 163, 130–136. [Google Scholar] [CrossRef]
- Kamm, U.; Rotach, P.; Gugerli, F.; Siroky, M.; Edwards, P.; Holderegger, R. Frequent long-distance gene flow in a rare temperate forest tree (Sorbus domestica) at the landscape scale. Heredity 2009, 103, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Nason, J.D.; Hamrick, J.L. Reproductive and genetic consequences of forest fragmentation: Two case studies of Neotropical canopy trees. J. Hered. 1997, 88, 264–276. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Elam, D.R. Population genetic consequences of small population size for plant conservation. Ann. Rev. Ecol. Syst. 1993, 24, 217–242. [Google Scholar] [CrossRef]
- Robledo-Arnuncio, J.J.; Alía, R.; Gil, L. Increased selfing and correlated paternity in a small population of a predominantly outcrossing conifer, Pinus sylvestris. Mol. Ecol. 2004, 13, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Lande, R. Genetics and demography in biological conservation. Science 1988, 241, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Aguinagalde, I.; Hampe, A.; Mohanty, A.; Martín, J.P.; Duminil, J.; Petit, R.J. Effects of life history traits and species distribution on genetic structure at maternally inherited markers in European trees and shrubs. J. Biogeogr. 2005, 32, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Müller-Starck, G.; Baradat, Ph.; Bergmann, F. Genetic variation within European tree species. New For. 1992, 6, 23–47. [Google Scholar] [CrossRef]
- Ennos, R.A. Estimating the relatives rates of pollen and seed migration among plant populations. Heredity 1994, 72, 250–259. [Google Scholar] [CrossRef]
- Oddou-Muratorio, S.; Petit, R.J.; Le Guerroué, B.; Guesnet, D.; Demesure, B. Pollen- versus seed-mediated gene flow in a scattered forest tree species. Evolution 2001, 55, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Bacles, C.F.E.; Lowe, A.J.; Ennos, R.A. Effective seed dispersal across a fragmented landscape. Science 2006, 311, 628. [Google Scholar] [CrossRef] [PubMed]
- Slavov, G.T.; DiFazio, S.P.; Strauss, S.H. Gene flow in forest trees: from empirical estimates to transgenic risk assessment. In Scientific Methods Workshop: Ecological and Agronomic Consequences of Gene Flow from Transgenic Crops to Wild Relatives; Snow, A., Mallory-Smith, C., Ellstrand, N., Holt, J., Quemada, H., Eds.; Ohio State University: Colombus, OH, USA, 2002; pp. 113–133. [Google Scholar]
- Bacles, C.F.E.; Ennos, R.A. Paternity analysis of pollen-mediated gene flow for Fraxinus excelsior L. in a chronically fragmented landscape. Heredity 2008, 101, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Harju, A.M.; Nikkanen, T. Reproductive success of orchard and nonorchard pollens during different stages of pollen shedding in a Scots pine seed orchard. Can. J. For. Res. 1996, 26, 1096–1102. [Google Scholar] [CrossRef]
- Robledo-Arnuncio, J.J.; Gil, L. Patterns of pollen dispersal in a small population of Pinus sylvestris L. revealed by total exclusion paternity analysis. Heredity 2005, 94, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Knowles, P. Mating system and effective pollen immigration in a Norway spruce (Picea abies (L.) Karst) plantation. Silvae Genet. 1994, 43, 48–52. [Google Scholar]
- Wang, K.S. Gene flow in European beech (Fagus sylvatica L.). Genetica 2004, 122, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Piotti, A.; Leonardi, S.; Buiteveld, J.; Geburek, T.; Gerber, S.; Kramer, K.; Vettori, C.; Vendramin, G.G. Comparison of pollen gene flow among four European beech (Fagus sylvatica L.) populations characterized by different management regimes. Heredity 2012, 108, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Thomasset, M.; Hodkinson, T.R.; Restoux, G.; Frascaria-Lacoste, N.; Douglas, G.C.; Fernandez-Manjarres, J.F. Thank you for not flowering: Conservation genetics and gene flow analysis of native and non-native populations of Fraxinus (Oleaceae) in Ireland. Heredity 2014, 112, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Pakkanen, A.; Nikkanen, T.; Pulkkinen, P. Annual variation in pollen contamination and outcrossing in a Picea abies seed orchard. Scand. J. For. Res. 2000, 15, 399–404. [Google Scholar]
- Burczyk, J.; Lewandowski, A.; Chalupka, W. Local pollen dispersal and distance gene flow in Norway spruce (Picea abies (L.) Karst.). For. Ecol. Manag. 2004, 197, 39–48. [Google Scholar] [CrossRef]
- Nikolic, D.; Tucic, N. Isoenzyme variation within and among populations of European black pine (Pinus nigra Arnold). Silvae Genet. 1983, 32, 80–89. [Google Scholar]
- Kinloch, B.B.; Westfall, R.D.; Forrest, G.I. Caledonian scots pine: origins and genetic structure. New Phytol. 1986, 104, 703–729. [Google Scholar] [CrossRef]
- Buiteveld, J.; Bakker, E.G.; Bovenschen, J.; de Vries, S.G.M. Paternity analysis in a seed orchard of Quercus robur L. and estimation of the amount of background pollination using microsatellite markers. For. Genet. 2001, 8, 331–337. [Google Scholar]
- Barton, N.H.; Slatkin, M. A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 1986, 56, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.F. Basic Concepts in Population, Quantitative, and Evolutionary Genetics; W.H. Freeman and Company: New York, NY, USA, 1986; p. 273. [Google Scholar]
- Klekowski, J. Genetic load and its causes in long-lived plants. Trees 1988, 2, 195–203. [Google Scholar] [CrossRef]
- Tinner, W.; Lotter, A.F. Holocene expansions of Fagus silvatica and Abies alba in central Europe: Where are we after eight decades of debate? Quat. Sci. Rev. 2006, 526–549. [Google Scholar] [CrossRef]
- Williams, C.G.; Savolainen, O. Inbreeding depression in conifers: Implications for breeding strategy. For. Sci. 1996, 42, 102–117. [Google Scholar]
- Sorensen, F.C. The roles of polyembryony and embryo viability in the genetic system of conifers. Evolution 1982, 36, 725–733. [Google Scholar] [CrossRef]
- Caballero, A. Developments in the prediction of effective population size. Heredity 1994, 73, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, A.I. Interpopulation gene flow in Hepatica nobilis and Corydalis cava at the border of their range. Zhurnal Obshchei Biol. 1997, 58, 84–93. [Google Scholar]
- Shao, J.-W.; Wang, J.; Xu, Y.-N.; Pan, Q.; Shi, Y.; Kelso, S.; Lv, G.-S. Genetic diversity and gene flow within and between two different habitats of Primula merrilliana (Primulaceae), an endangered distylous forest herb in eastern China. Bot. J. Linn. Soc. 2015, 179, 172–189. [Google Scholar] [CrossRef]
- Amler, K.; Bahl, A.; Henle, K.; Kaule, G.; Poschlod, P.; Settele, J. (Eds.) Populationsbiologie in der Naturschutzpraxis. Isolation, Flächenbedarf und Biotopansprüche von Pflanzen und Tieren; Ulmer: Stuttgart, Germany, 1999; p. 336.
- Aavik, T.; Holderegger, R.; Edwards, P.J.; Billeter, R. Patterns of contemporary gene flow suggest low functional connectivity of grasslands in a fragmented agricultural landscape. J. Appl. Ecol. 2013, 50, 395–403. [Google Scholar] [CrossRef]
- Stebbins, G.L. Flowering Plants: Evolution above the Species Level; Harvard University Press: Cambridge, MA, USA, 1974. [Google Scholar]
- Godt, M.J.W.; Hamrick, J.L. Patterns and levels of pollen-mediated gene flow in Lathyrus latifolius. Evolution 1993, 47, 98–110. [Google Scholar] [CrossRef]
- Fischer, M.A.; Adler, W.; Oswald, K. Exkursionsflora für Österreich, Lichtenstein und Südtirol, 3rd ed.; Biologiezentrum des Oberösterreichischen Landesmuseums: Linz, Austria, 2008; p. 1392. [Google Scholar]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: Fst and related measures. Mol. Ecol. Res. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Krutovskii, K.V.; Erofeeva, S.Y.; Aagaard, J.E.; Strauss, S.H. Simulation of effects of dominance on estimates of population genetic diversity and differentiation. J. Hered. 1999, 90, 499–502. [Google Scholar] [CrossRef]
- Jost, L. Gst and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Weiss, G.H. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 1964, 49, 561–576. [Google Scholar] [PubMed]
- Adams, W.T. Gene dispersal within forest tree populations. New For. 1992, 6, 217–240. [Google Scholar] [CrossRef]
- Lanner, R.M. Needed: A new approach to the study of pollen dispersion. Silvae Genet. 1966, 15, 50–52. [Google Scholar]
- Silen, R.R. Pollen dispersal consideration for Douglas-fir. J. For. 1962, 60, 790–795. [Google Scholar]
- Dick, C.W. Genetic rescue of remnant tropical trees by an alien pollinator. Proc. R. Soc. Biol. Sci. Ser. B 2001, 268, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- White, G.M.; Boshier, D.H.; Powell, W. Increased pollen flow counteracts fragmentation in a tropical dry forest: An example from Swietenia humilis Zuccarini. Proc. Natl. Acad. Sci. USA 2002, 99, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Robledo-Arnuncio, J.J.; Smouse, P.E.; Gil, L.; Alia, R. Pollen movement under alternative silvicultural practices in native populations of Scots pine (Pinus sylvestris L.) in central Spain. For. Ecol. Manag. 2014, 197, 245–255. [Google Scholar] [CrossRef]
- Kimura, M.; Crow, J.F. The measurement of effective population numbers. Evolution 1963, 17, 279–288. [Google Scholar] [CrossRef]
- Klotz, S.; Kühn, I.; Durka, W. Biolflor—Eine Datenbank mit biologisch-ökologischen Merkmalen zur Flora von Deutschland. Schrift. Vegetationskunde 2002, 38, 1–334. [Google Scholar]
- Rohmeder, E. Das Saatgut in der Forstwirtschaft; Paul Parey: Hamburg, Germany; Berlin, Germany, 1972; p. 273. [Google Scholar]
- Hintze, C.; Heydel, F.; Hoppe, C.; Cunze, S.; König, A.; Tackenberg, O. D3: The dispersal and diaspore database—Baseline data and statistics on seed dispersal. Perspect. Plant Ecol. 2013, 15, 180–192. [Google Scholar] [CrossRef]
- The Tree of Sex Consortium. Tree of Sex: A database of sexual systems. Sci. Data 2014, 1, 140015. [Google Scholar]
- Yampolsky, C.; Yampolsky, H. Distribution of sex forms in the phanaerogamic flora. Bibliogr. Genet. 1922, 3, 1–62. [Google Scholar]
- Barton, L.V. Seed Preservation and Longevity; Hill: London, UK, 1961. [Google Scholar]
- Baskin, C.C.; Baskin, J.M. Seeds. Ecology, Biogeography, and Evolution of Dormancy and Germination; Academic Press: Amsterdam, The Netherlands, 2014; p. 1600. [Google Scholar]
- Flather, C.H.; Hayward, G.D.; Beissinger, S.R.; Stephens, P.A. Minimum viable populations: Is there a ‘magic number’ for conservation practitioners? Trends Ecol. Evol. 2011, 26, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Balloux, F. EASYPOP (version 1.7): A computer program for population genetics simulations. J. Hered. 2001, 92, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Heckel, G. Computer programs for population genetics data analysis: A survival guide. Nat. Rev. Genet. 2006, 7, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Pelikan, S.; Rogstad, S.H. NEWGARDEN: A computer program to model the population dynamics and genetics of establishing and fragmented plant populations. Conserv. Genet. Res. 2013, 5, 857–862. [Google Scholar] [CrossRef]
- Machon, N.; Baradat, D.; Godelle, B. METAPOP a Program Simulating Evolutionary Processes Acting on Metapopulations; Laboratoire ES, Paris-Sud: Paris, France, 1995. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of Inheritance | N | Median | Min | Max | 1 | 2 |
|---|---|---|---|---|---|---|
| biparental (1) | 231 | 4.2 | 0.2 | 55.0 | ||
| paternal (2) | 15 | 7.0 | 2.0 | 30.8 | n.s. | |
| maternal (3) | 85 | 61.0 | 1.2 | 103.4 | *** | *** |
| Mode of Pollen Dispersal | N | Median | Min | Max | 1 | 2 |
|---|---|---|---|---|---|---|
| wind-pollinated (1) | 157 | 3.80 | 0.2 | 32.0 | ||
| insect-pollinated (2) | 20 | 5.85 | 1.4 | 22.8 | . | |
| mixed (3) | 21 | 8.70 | 0.8 | 33.0 | ** | n.s. |
| Mode of Diaspore Dispersal | N | Median | Min | Max | 1 | df |
|---|---|---|---|---|---|---|
| biparentally inherited | ||||||
| anemochorous (1) | 94 | 4.1 | 0.2 | 33 | ||
| endozoochorous (2) | 104 | 4.6 | 0.3 | 32 | n.s. | 194 |
| maternally inherited | ||||||
| anemochorous (1) | 49 | 59.60 | 1.7 | 100.0 | ||
| endozoochorous (2) | 36 | 62.45 | 1.2 | 103.4 | n.s. | 81 |
| Species | Distance (m) | Gene Flow | Method | References |
|---|---|---|---|---|
| Fagus sylvatica | >300/>500 | 0.7/0.9 a | paternity exclusion | [78] |
| n.a. | 52.6–79.8 a | paternity exclusion | [79] | |
| Fraxinus excelsior | >600 | 46–95 a | paternity exclusion | [6] |
| n.a. | 43–68 a | paternity analysis | [74] | |
| <100/300–1900/2900 | 85/15/<1 a | |||
| <100/>400 | <35/>30 a | paternity analysis | [80] | |
| Picea abies | n.a. | 70 a | paternity exclusion | [81] b |
| >4,000 c | 16 a | paternity exclusion | [77] b | |
| >100 | 83 a | paternity exclusion | [82] | |
| Pinus nigra | n.a. | 21.83 d/4.15 e | indirect | [83] b |
| Pinus sylvestris | >2000 c | 48 a | paternity exclusion | [75] b |
| n.a. | 4.55 d/8.05 e | indirect | [84] b | |
| ≥30,000 | 4.3 a | paternity exclusion | [76] | |
| Quercus petraea | n.a. | 69 a | paternity exclusion | [31] b |
| Quercus robur | n.a. | 65 a | paternity exclusion | [31] b |
| >400 c | 70 a | paternity exclusion | [85] b | |
| Quercus robur and Q. petraea g | n.a. | 20.9–81.1 a | paternity exclusion | [37] |
| Quercus petraea and Q. pubescens g | n.a. | 39.2 a | paternity exclusion | [37] |
| Sorbus domestica | 1000/3000/12,000–16,000 | >33/8/1.8 a | paternity analysis | [62,63] |
| Sorbus torminalis | >1000 c | 1.34/2.85 f | paternity analysis | [35] |
| Life History Traits (Intrinsic) | Unfavorable States | Expected Effects |
|---|---|---|
| mode of pollen transfer 1 | animal-pollinated | • lower dispersal abilities compared to wind-pollinated species |
| species relying on specialized pollinators | • reduced pollination and possibly occurrence of pollen limitation in case of low population densities or activities of pollinators | |
| mode of diaspore transfer 2 | barochor, vectors of low mobility | • reduced gene flow due to low dispersal abilities of diaspores |
| breeding system 3 | dioecy, asymmetric distribution of sexes | • increased random genetic drift due to lower effective population size |
| mating system (selfing, outcrossing, apomixis) 4 | selfing; apomixis | |
| life form 5 | annual, short-lived | • random genetic drifts act quickly due to short generation times |
| durability of the seed bank 6 | low | • genetic bottleneck effects (i.e., random genetic drift) in case of fluctuating seed set |
| ploidy 7 | diploidy | • lower effective population size and lower in tendency reduced heterozygosity compared to polyploids (polysomatic inheritance provided) |
| Population characteristics (extrinsic) | ||
| size and density (habitat quality) | low | • high random genetic drift connected to low effective population size |
| • reduced seed set due to insufficient supply of resources needed by pollinators | ||
| • reduced gene flow through pollen and/or diaspores due to low emission by potential source populations | ||
| population distances | high | • distance reduces the probability of pollen and diaspore immigration |
| type of barrier to gene flow | strong physical barrier for non-volant or poorly volant vectors; corridors of changed environment hampering movement of stenoecious vectors | • reduced or blocked gene flow |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobeš, C.; Konrad, H.; Geburek, T. Potential Population Genetic Consequences of Habitat Fragmentation in Central European Forest Trees and Associated Understorey Species—An Introductory Survey. Diversity 2017, 9, 9. https://doi.org/10.3390/d9010009
Dobeš C, Konrad H, Geburek T. Potential Population Genetic Consequences of Habitat Fragmentation in Central European Forest Trees and Associated Understorey Species—An Introductory Survey. Diversity. 2017; 9(1):9. https://doi.org/10.3390/d9010009
Chicago/Turabian StyleDobeš, Christoph, Heino Konrad, and Thomas Geburek. 2017. "Potential Population Genetic Consequences of Habitat Fragmentation in Central European Forest Trees and Associated Understorey Species—An Introductory Survey" Diversity 9, no. 1: 9. https://doi.org/10.3390/d9010009