Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Genomic DNA Isolation
2.2. Analysis of SSR Markers
2.3. Marker Data Analysis
3. Results
3.1. Descriptive Statistics of SSR Marker Loci
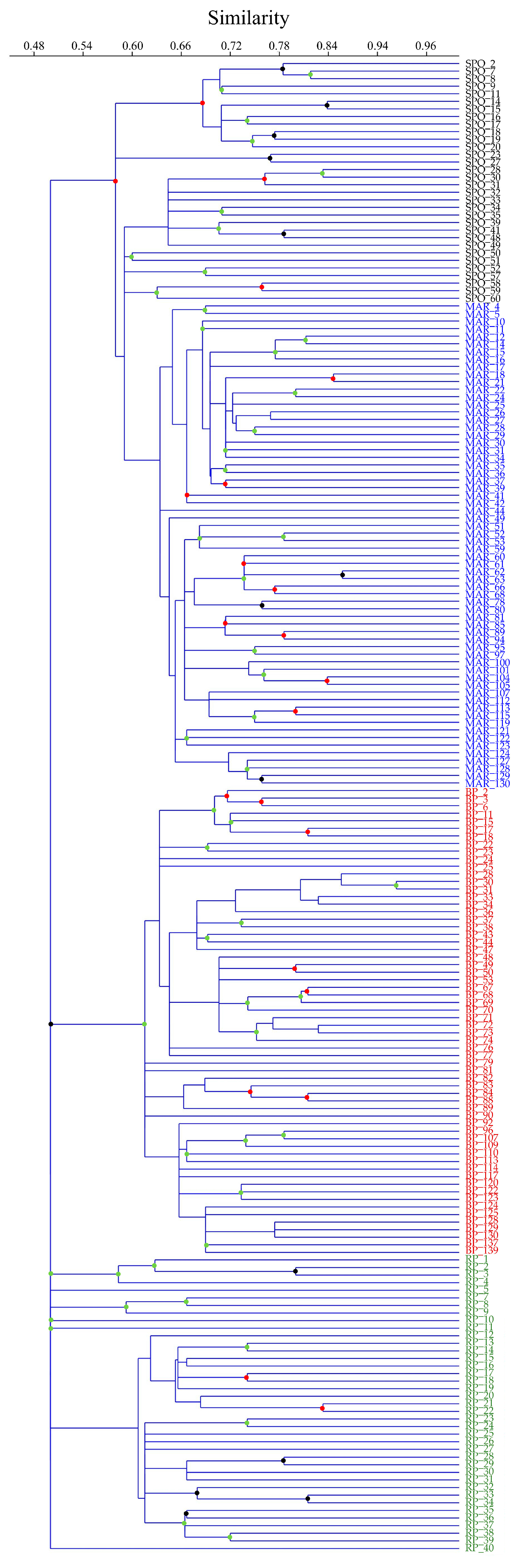
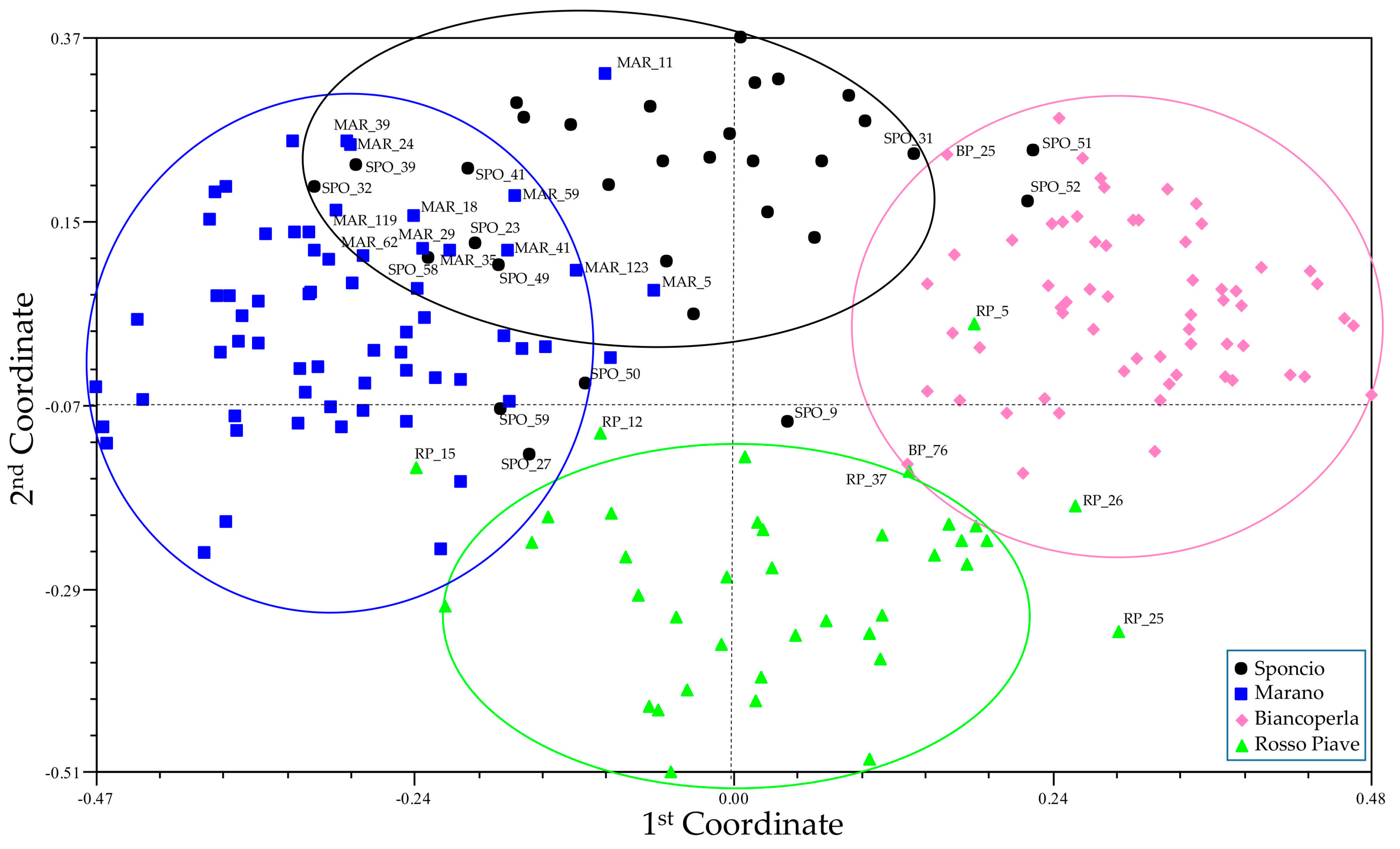
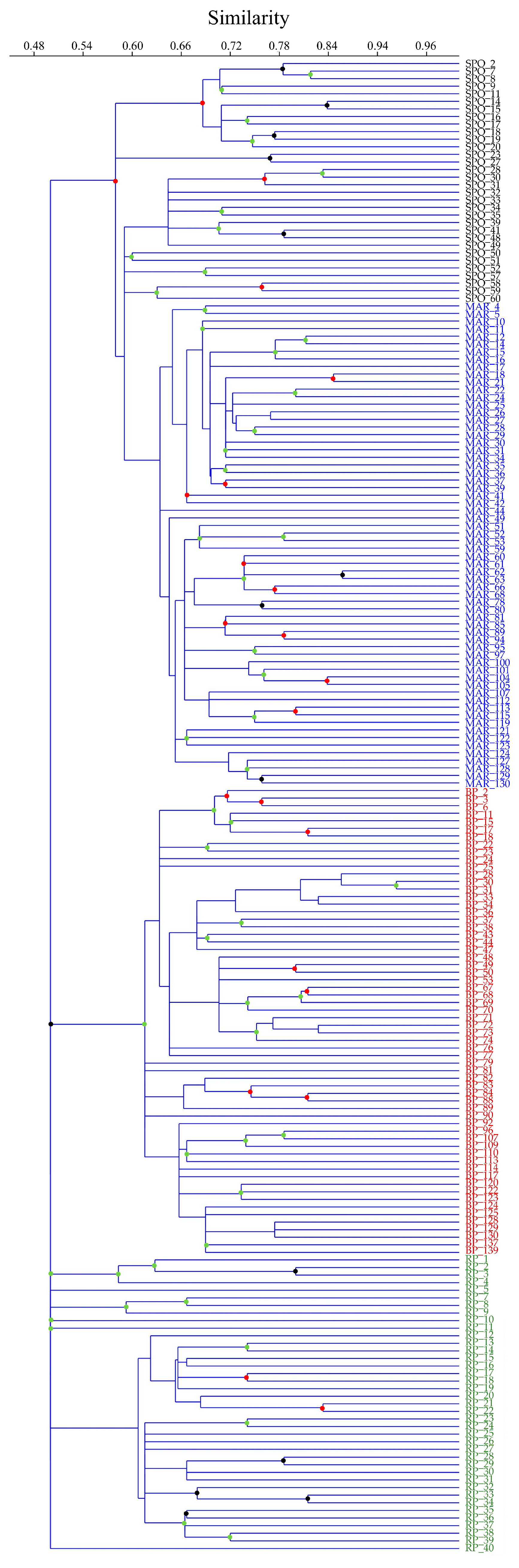
3.2. Genetic Diversity and Cluster Analysis
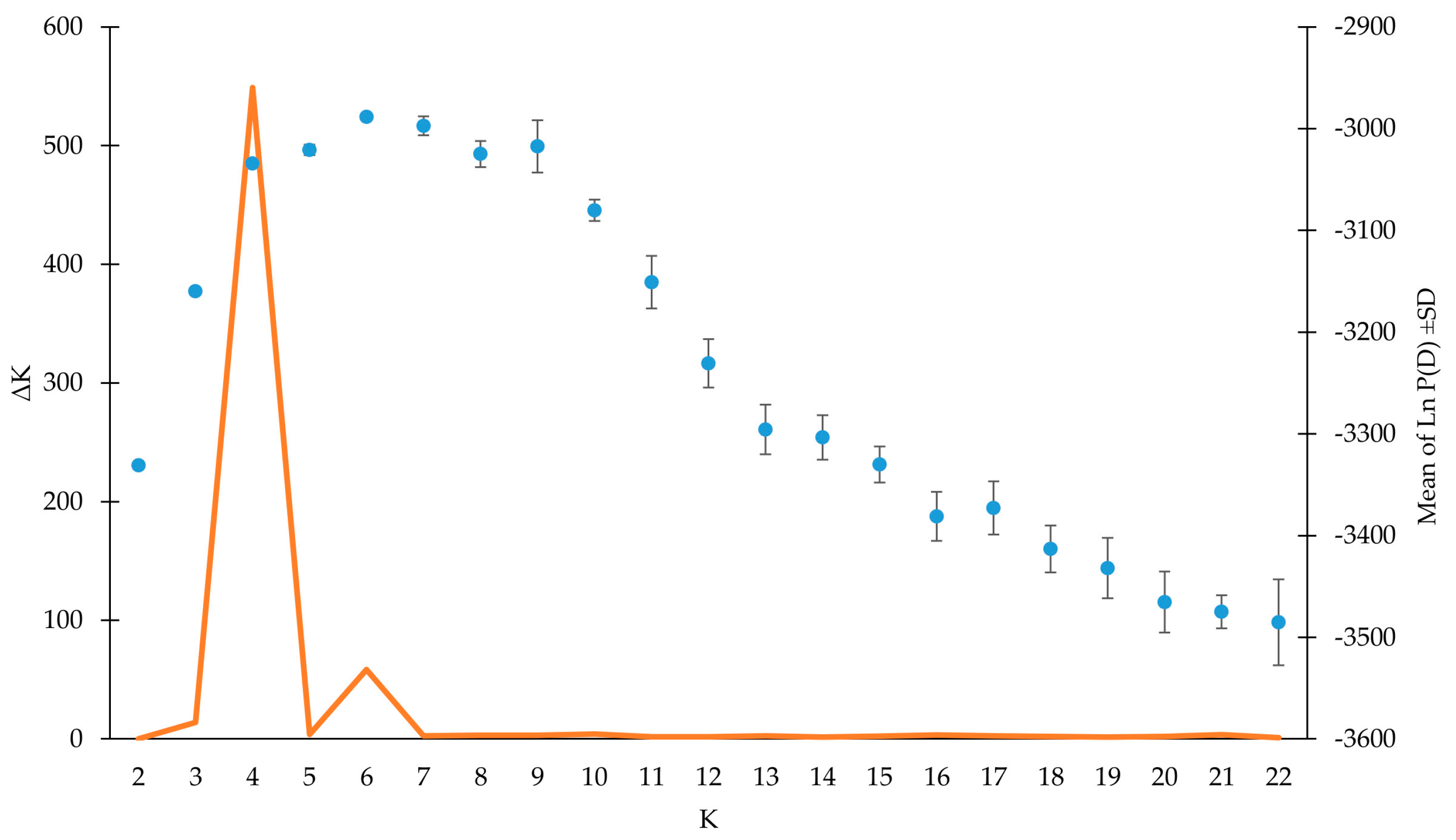
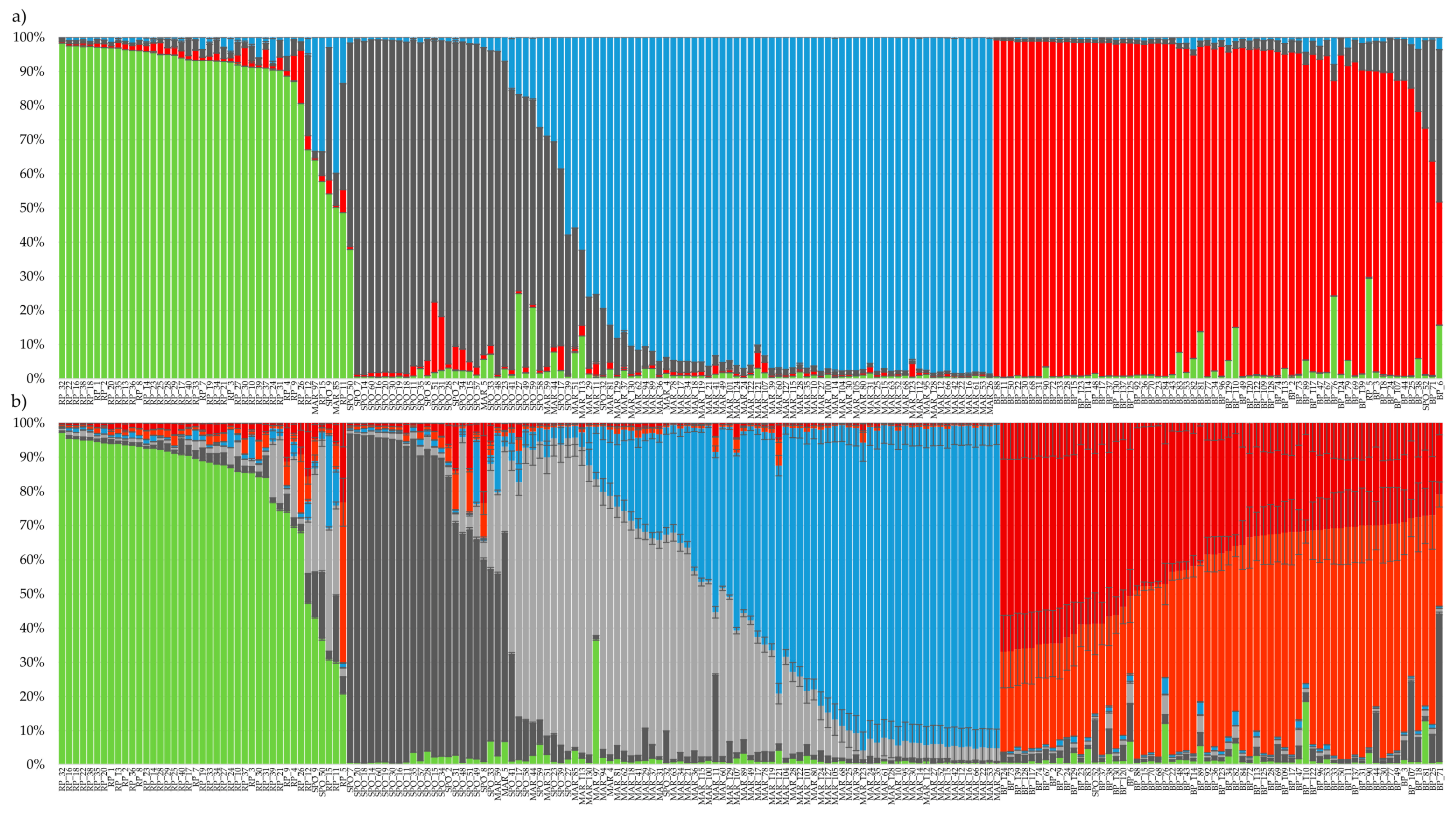
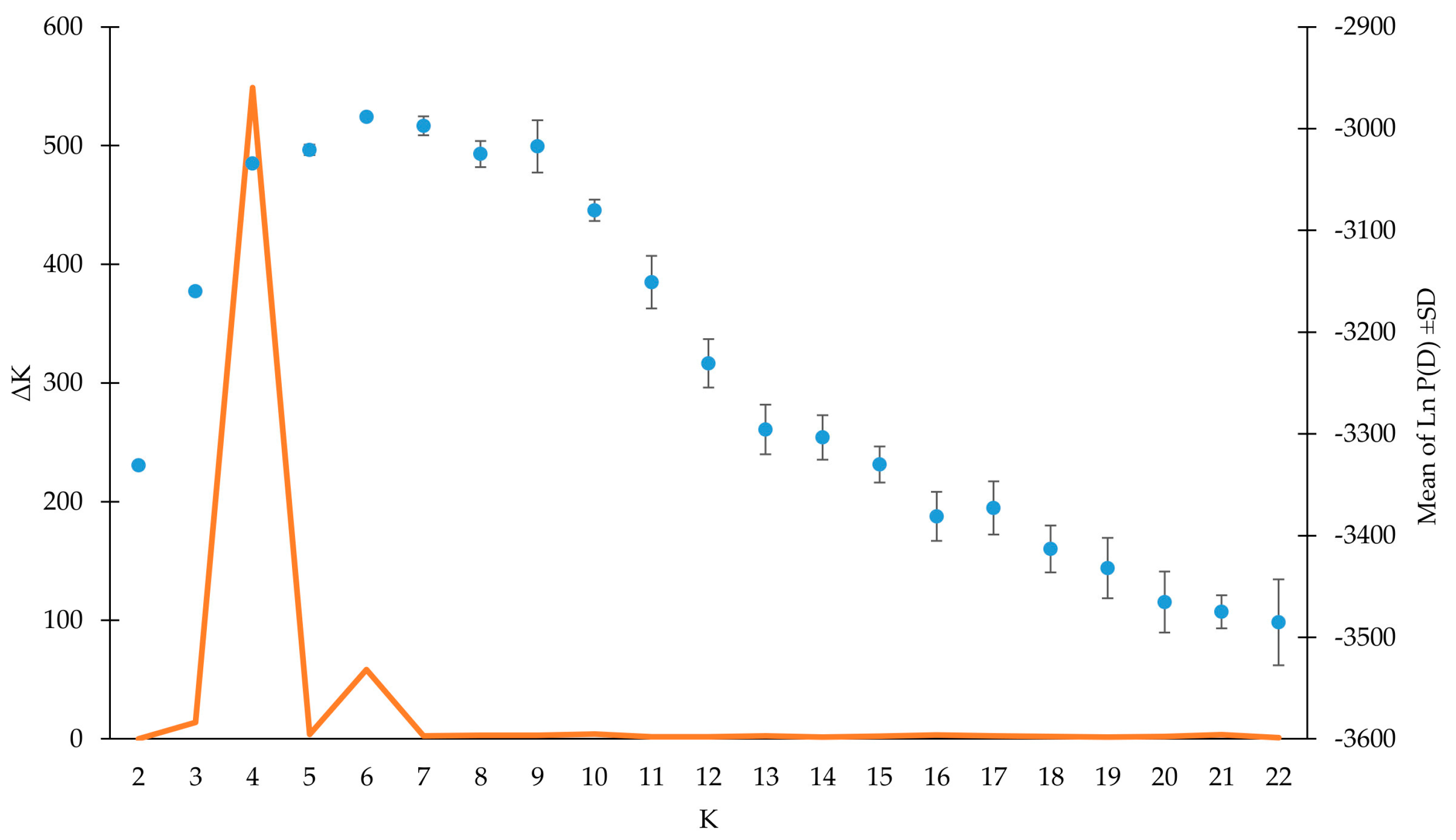
3.3. Genetic Structure Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baur, E. Die Bedeutung der primitiven Kulturrassen und der wilden Verwandten unserer Kulturpflanzen fur die Pflanzenzuchtung. Jahrb. DLG 1914, 21, 104–110. [Google Scholar]
- Frankel, O.; Bennett, E. Genetic Resources in Plants—Their Exploration and Conservation (IBP Handbook No. 11); Blackwell Science Ltd.: London, UK, 1970. [Google Scholar]
- Harlan, J. Our vanishing genetic resources. Science 1975, 188, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Brush, S. In situ conservation of landraces in centers of crop diversity. Crop Sci. 1995, 35, 346–354. [Google Scholar] [CrossRef]
- Bellon, M.R. Conceptualizing interventions to support on-farm genetic resource conservation. World Dev. 2004, 32, 159–172. [Google Scholar] [CrossRef]
- Lucchin, M.; Barcaccia, G.; Parrini, P. Characterization of a flint maize (Zea mays L. convar. mays) Italian landrace: I. Morpho-phenological and agronomic traits. Genet. Resour. Crop Evol. 2003, 50, 315–327. [Google Scholar] [CrossRef]
- Eurostat: Crop Statistics. Available online: http://ec.europa.eu/eurostat/web/agriculture/data/database?p_p_id=NavTreeportletprod_WAR_NavTreeportletprod_INSTANCE_ff6jlD0oti4U&p_p_lifecycle=0&p_p_state=normal&p_p_mode=view&p_p_col_id=column-2&p_p_col_pos=1&p_p_col_count=2 (accessed on 31 July 2017).
- Shanbao, Q.; Yuhua, W.; Tingzhao, R.; Kecheng, Y.; Shibin, G.; Guangtang, P. Effective improvement of genetic variation in maize lines derived from R08xDonor backrosses by SSRs. Biotechnology 2009, 8, 358–364. [Google Scholar] [CrossRef]
- Cömertpay, G.; Baloch, F.S.; Kilian, B.; Ülger, A.C.; Özkan, H. Diversity assessment of Turkish maize landraces based on fluorescent labelled SSR markers. Plant Mol. Biol. Report. 2012, 30, 261–274. [Google Scholar] [CrossRef]
- Pineda-Hidalgo, K.V.; Méndez-Marroquín, K.P.; Alvarez, E.V.; Chávez-Ontiveros, J.; Sánchez-Peña, P.; Garzón-Tiznado, J.A.; Vega-García, M.O.; López-Valenzuela, J.A. Microsatellite-based genetic diversity among accessions of maize landraces from sinaloa in México. Hereditas 2013, 150, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Qi-Lun, Y.; Ping, F.; Ke-Cheng, K.; Guang-Tang, P. Genetic diversity based on SSR markers in maize (Zea mays L.) landraces from Wuling mountain region in China. J. Genet. 2008, 87, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Oppong, A.; Bedoya, C.A.; Ewool, M.B.; Asante, M.D.; Thomson, R.N.; Adu-Dapaah, H.; Lamptey, J.N.; Ofori, K.; Offei, S.K.; Warburton, M.L. Bulk genetic characterization of Ghanaian maize landraces using microsatellite markers. Maydica 2014, 59, 1–8. [Google Scholar]
- Barcaccia, G.; Lucchin, M.; Parrini, P. Characterization of a flint maize (Zea mays var. indurata) Italian landrace, II. Genetic diversity and relatedness assessed by SSR and Inter-SSR molecular markers. Genet. Resour. Crop Evol. 2003, 50, 253–271. [Google Scholar] [CrossRef]
- Mais Sponcio—Cooperativa Agricola La Fiorita. Available online: http://www.cooperativalafiorita.it/?scheda_prodotto+prodotti=mais_sponcio (accessed on 30 June 2017).
- Perco, D. Antonio Maresio Bazolle/Il Possidente Bellunes; Tip. B. Bernardino: Feltre, Italy, 1986. [Google Scholar]
- Zapparoli, T.V. Il Granoturco Marano. L’Italia Agric. 1939, 76, 155–159. [Google Scholar]
- Zaccaria, L. Valutazione delle Variazioni dei Caratteri Morfo-Fisiologici Intervenute nel Corso Degli Anni Nella Varietà di Mais Marano Vicentino. Bachelor’s Thesis, University of Padua, Padua, Italy, 31 Octorber 2012. [Google Scholar]
- Bernardi, U.; Demattè, E. Giacomo Agostinetti/Cento e Dieci ricordi che Formano il Buon Fattor di Villa; Neri Pozza: Pordenone, Italy, 1998. [Google Scholar]
- Miniscalco, V. Il Granoturco; Arti grafiche F.lli Cosarini: Pordenone, Italy, 1946. [Google Scholar]
- Clemens, R.L.B. Keeping Farmers on the Land: Adding Value in Agriculture in the Veneto Region of Italy. MATRIC Briefing Papers 6. 2004. Available online: http://lib.dr.iastate.edu/matric_briefingpapers/6 (accessed on 13 August 2017).
- Cassidy, A.; Mukamal, K.J.; Liu, L.; Franz, M.; Eliassen, A.H.; Rimm, E.B. High anthocyanin intake is associated with a reduced risk of myocardial infarction in young and middle-aged women. Circulation 2013, 127, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments A poor man’s approach to genotyping for research and high-throughput diagnostics. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Register, J.I.; Sullivan, H.; Yun, Y.; Cook, D.; Vaske, D. A set of microsatellite markers of general utility in maize. Maize Genet. Coop. News Lett. 2001, 75, 31–34. [Google Scholar]
- Nagy, S.; Poczai, P.; Cernák, I.; Gorji, A.M.; Hegedus, G.; Taller, J. PICcalc: An online program to calculate polymorphic information content for molecular genetic studies. Biochem. Genet. 2012, 50, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.C.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.H.; Mao, J.X. POPGENE, the User Friendly Shareware for Population Genetic Analysis; Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada, 1997. [Google Scholar]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Van Zonneveld, M.; Scheldeman, X.; Escribano, P.; Viruel, M.A.; van Damme, P.; Garcia, W.; Tapia, C.; Romero, J.; Sigueñas, M.; Hormaza, J.I. Mapping genetic diversity of cherimoya (Annona cherimola Mill.): Application of spatial analysis for conservation and use of plant genetic resources. PLoS ONE 2012, 7, e29845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Barcaccia, G.; Felicetti, M.; Galla, G.; Capomaccio, S.; Cappelli, K.; Albertini, E.; Buttazzoni, L.; Pieramati, C.; Silvestrelli, M.; Verini Supplizi, A. Molecular analysis of genetic diversity, population structure and inbreeding level of the Italian Lipizzan horse. Livest. Sci. 2013, 151, 124–133. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System, Version 2.1; Applied Biostatistics Inc.: Port Jefferson, NY, USA, 2004. [Google Scholar]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: PAleontological STatistics software package for education and data Analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, A.; Lareu, M.V. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Palumbo, F.; Galla, G.; Barcaccia, G. Developing a molecular identification assay of old landraces for the genetic authentication of typical agro-food products: The case study of the barley ‘Agordino’. Food Technol. Biotechnol. 2017, 55, 29–39. [Google Scholar] [CrossRef]
- Russell, W.A. Genetic improvement of maize yields. Adv. Agron. 1991, 46, 245–298. [Google Scholar] [CrossRef]
- Mais Biancoperla. Available online: http://www.legambientepadova.it/files/Scheda%20mais.pdf (accessed on 30 June 2017).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Locus ID | Motif | Size (bp) | Primer | Tm (°C) | PIC |
|---|---|---|---|---|---|---|
| 1 | phi056 | GCC | 103–112 | M13-ACGCCCAGATCTGTTCCTTCTC | 63 | 0.67 |
| ATGGCGGCAGGCCGATTGTT | ||||||
| 2 | phi127 | AGAC | 129–145 | M13-ATATGCATTGCCTGGAACTGGAAGGA | 62 | 0.70 |
| AATTCAAACACGCCTCCCGAGTGT | ||||||
| 3 | phi073 | CAG | 107–116 | M13-TTACTCCTATCCACTGCGGCCTGGAC | 69 | 0.65 |
| GCGGCATCCCGTACAGCTTCAGA | ||||||
| 4 | phi076 | GAGCGG | 182–192 | M13-TTCTTCCGCGGCTTCAATTTGACC | 61 | 0.65 |
| GCATCAGGACCCGCAGAGTC | ||||||
| 5 | phi024 | CCT | 183–195 | ACTGTTCCACCAAACCAAGCCGAGA | 69 | 0.69 |
| M13-AGTAGGGGTTGGGGATCTCCTCC | ||||||
| 6 | phi031 | GTAC | 174–177 | M13-GCAACAGGTTACATGAGCTGACGA | 66 | 0.57 |
| CCAGCGTGCTGTTCCAGTAGTT | ||||||
| 7 | phi057 | GCC | 211–215 | M13-CTCATCAGTGCCGTCGTCCAT | 66 | 0.61 |
| CAGTCGCAAGAAACCGTTGCC | ||||||
| 8 | umc1075 | ATTGC | 156–166 | M13-GAGAGATGACAGACACATCCTTGG | 57 | 0.69 |
| ACATTTATGATACCGGGAGTTGGA | ||||||
| 9 | phi016 | GGT | 173–176 | M13-TTCCATCATTGATCCGGGTGTCG | 60 | 0.52 |
| AAGGAGCAACATCCCATCCAGGAA | ||||||
| 10 | phi084 | GAA | 174–178 | M13-AGAAGGAATCCGATCCATCCAAGC | 59 | 0.49 |
| CACCCGTACTTGAGGAAAACCC |
| Locus | Na | Ne | PIC | I | Ho | He | FIS | FIT | FST | Nm |
|---|---|---|---|---|---|---|---|---|---|---|
| phi024 | 5.00 | 3.54 | 0.66 | 1.32 | 0.59 | 0.72 | 0.07 | 0.19 | 0.13 | 1.69 |
| phi127 | 6.00 | 2.24 | 0.47 | 0.95 | 0.43 | 0.55 | 0.12 | 0.22 | 0.11 | 1.97 |
| phi084 | 2.00 | 1.68 | 0.32 | 0.60 | 0.29 | 0.41 | −0.03 | 0.26 | 0.28 | 0.65 |
| phi076 | 3.00 | 2.95 | 0.59 | 1.09 | 0.48 | 0.66 | 0.14 | 0.24 | 0.12 | 1.84 |
| phi031 | 2.00 | 1.75 | 0.34 | 0.62 | 0.39 | 0.43 | 0.04 | 0.09 | 0.05 | 4.87 |
| phi057 | 4.00 | 2.72 | 0.55 | 1.07 | 0.48 | 0.63 | 0.01 | 0.27 | 0.26 | 0.70 |
| phi056 | 4.00 | 1.86 | 0.38 | 0.77 | 0.29 | 0.46 | −0.08 | 0.37 | 0.42 | 0.34 |
| phi073 | 3.00 | 2.94 | 0.59 | 1.09 | 0.55 | 0.66 | 0.01 | 0.17 | 0.17 | 1.24 |
| phi016 | 2.00 | 1.00 | 0.37 | 0.69 | 0.250 | 0.50 | 0.34 | 0.47 | 0.19 | 1.04 |
| umc1075 | 5.00 | 4.02 | 0.71 | 1.47 | 0.53 | 0.75 | 0.16 | 0.26 | 0.12 | 1.84 |
| All loci | 3.60 | 2.57 | 0.50 | 0.97 | 0.43 | 0.58 | 0.08 | 0.25 | 0.18 | 1.62 |
| St. dev. | 1.43 | 0.80 | 0.14 | 0.30 | 0.12 | 0.12 | 0.04 | 0.03 | 0.03 | 0.40 |
| Sponcio | 3.00 | 2.09 | na | 0.80 | 0.46 | 0.49 | 0.06 | 0.21 | 0.16 | 1.34 |
| Marano | 2.50 | 2.08 | na | 0.74 | 0.46 | 0.47 | 0.02 | 0.21 | 0.19 | 1.04 |
| Biancoperla | 2.50 | 1.90 | na | 0.69 | 0.37 | 0.44 | 0.15 | 0.35 | 0.24 | 0.78 |
| Rosso Piave | 3.30 | 2.30 | na | 0.89 | 0.45 | 0.53 | 0.16 | 0.23 | 0.08 | 3.01 |
| All landraces | 2.82 | 2.09 | na | 0.80 | 0.43 | 0.48 | 0.10 | 0.25 | 0.17 | 1.54 |
| St. dev. | 0.39 | 0.16 | na | 0.09 | 0.04 | 0.04 | 0.07 | 0.07 | 0.07 | 1.00 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, F.; Galla, G.; Martínez-Bello, L.; Barcaccia, G. Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production. Diversity 2017, 9, 32. https://doi.org/10.3390/d9030032
Palumbo F, Galla G, Martínez-Bello L, Barcaccia G. Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production. Diversity. 2017; 9(3):32. https://doi.org/10.3390/d9030032
Chicago/Turabian StylePalumbo, Fabio, Giulio Galla, Liliam Martínez-Bello, and Gianni Barcaccia. 2017. "Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production" Diversity 9, no. 3: 32. https://doi.org/10.3390/d9030032