Determination of Anthracene on Ag-Au Alloy Nanoparticles/Overoxidized-Polypyrrole Composite Modified Glassy Carbon Electrodes



Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents and Materials
2.2. Measurement and Instrumentation
2.3. Preparation of Monometallic Silver Nanoparticles, Gold Nanoparticles and Silver-Gold Alloy Nanoparticles
2.4. Characterization of the Ag-Au Bimetallic Nanoparticles
2.5. Fabrication of Over-Oxidized Polypyrrole/Ag-AuNPs Composite Film-Modified Electrode (PPyox/Ag-AuNPs/GCE)
3. Results and Discussion
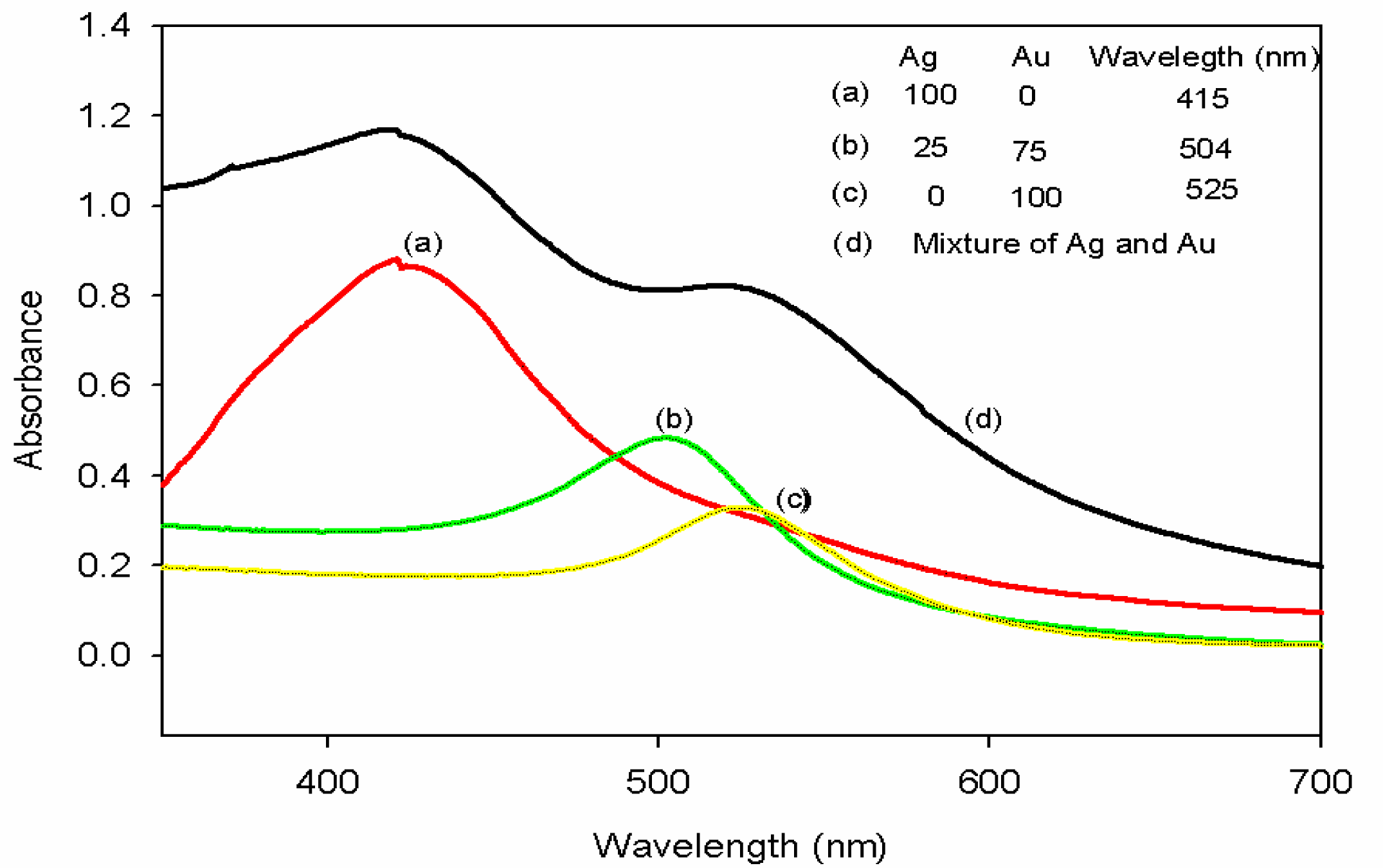
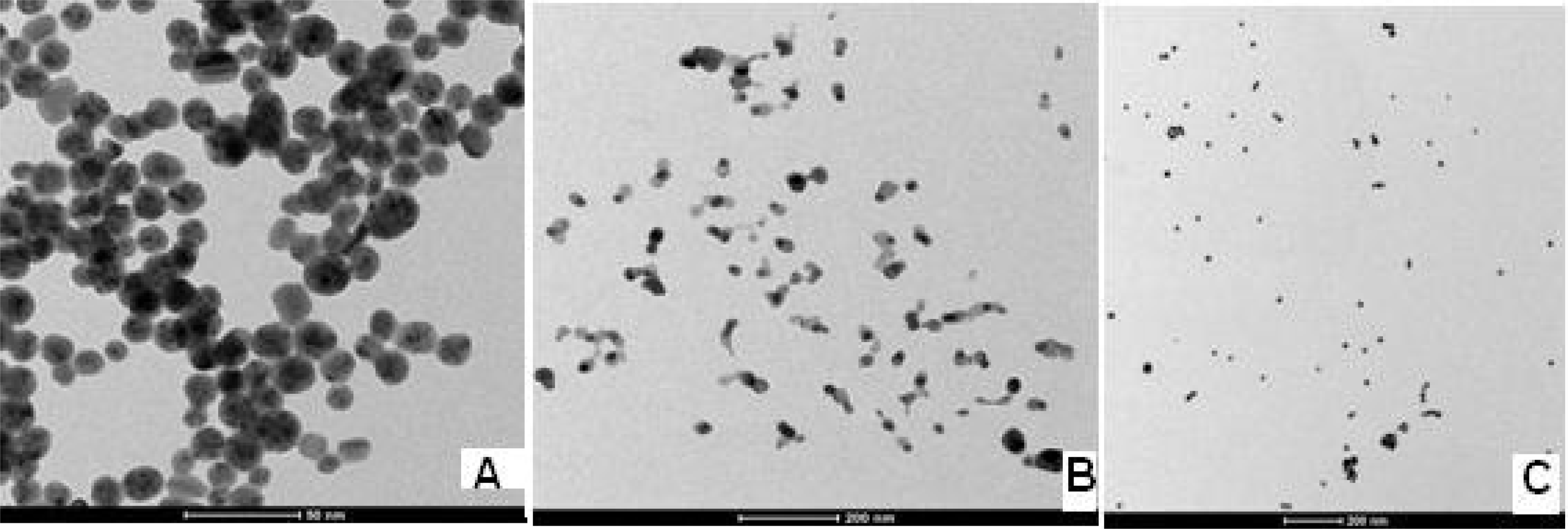
3.1. Characterization of Silver-Gold Alloy Nanoparticles
3.2. Fluorescence Properties of Silver-Gold Bimetallic Alloy Nanoparticles
3.3. Electrochemical Characterization of Ag-Au Alloy Nanoparticles and PPyox/Ag-AuNPs/GCE
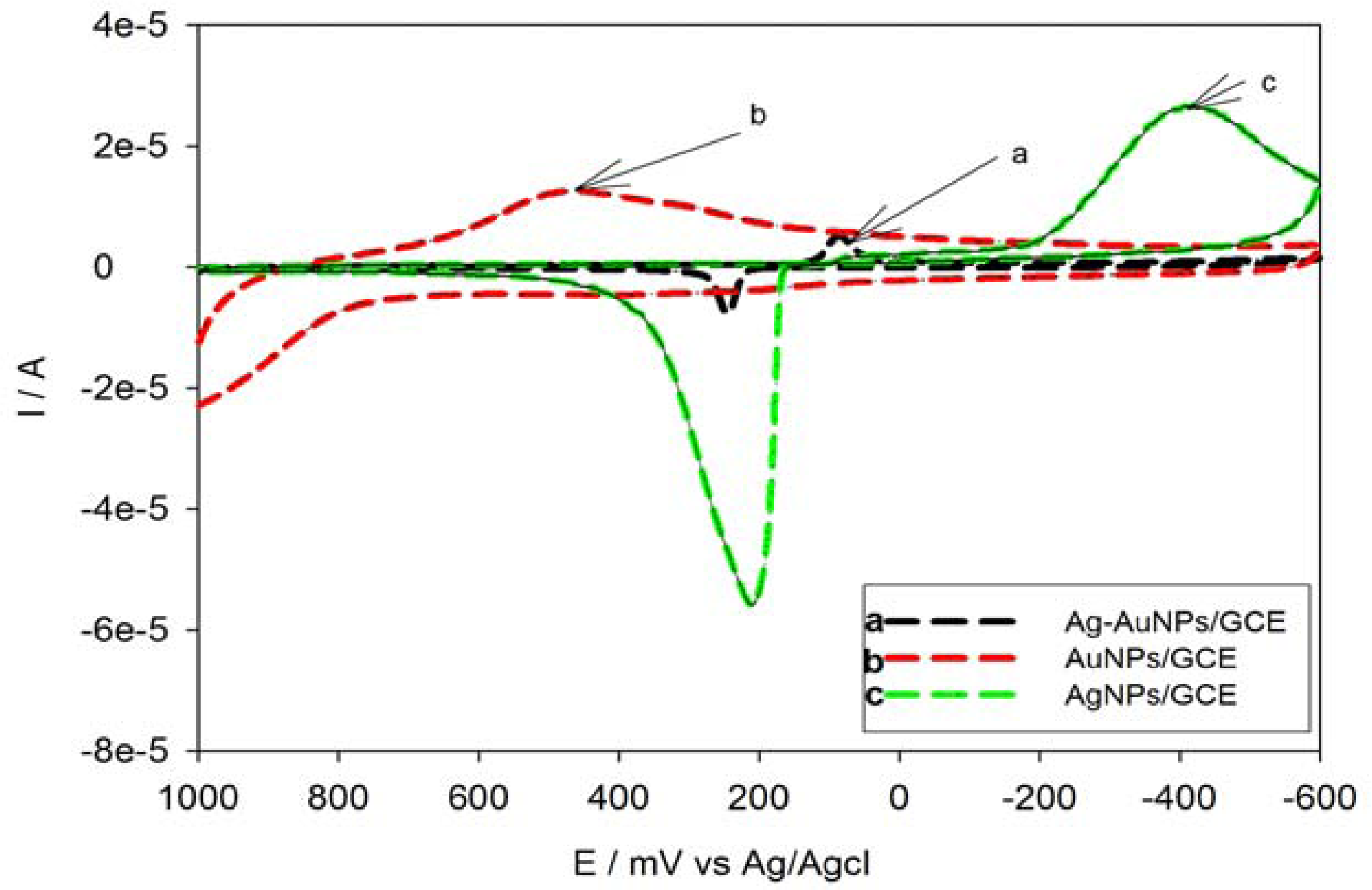
3.3.1. Characterization of Ag-AuNPs/GCE in Neutral Medium
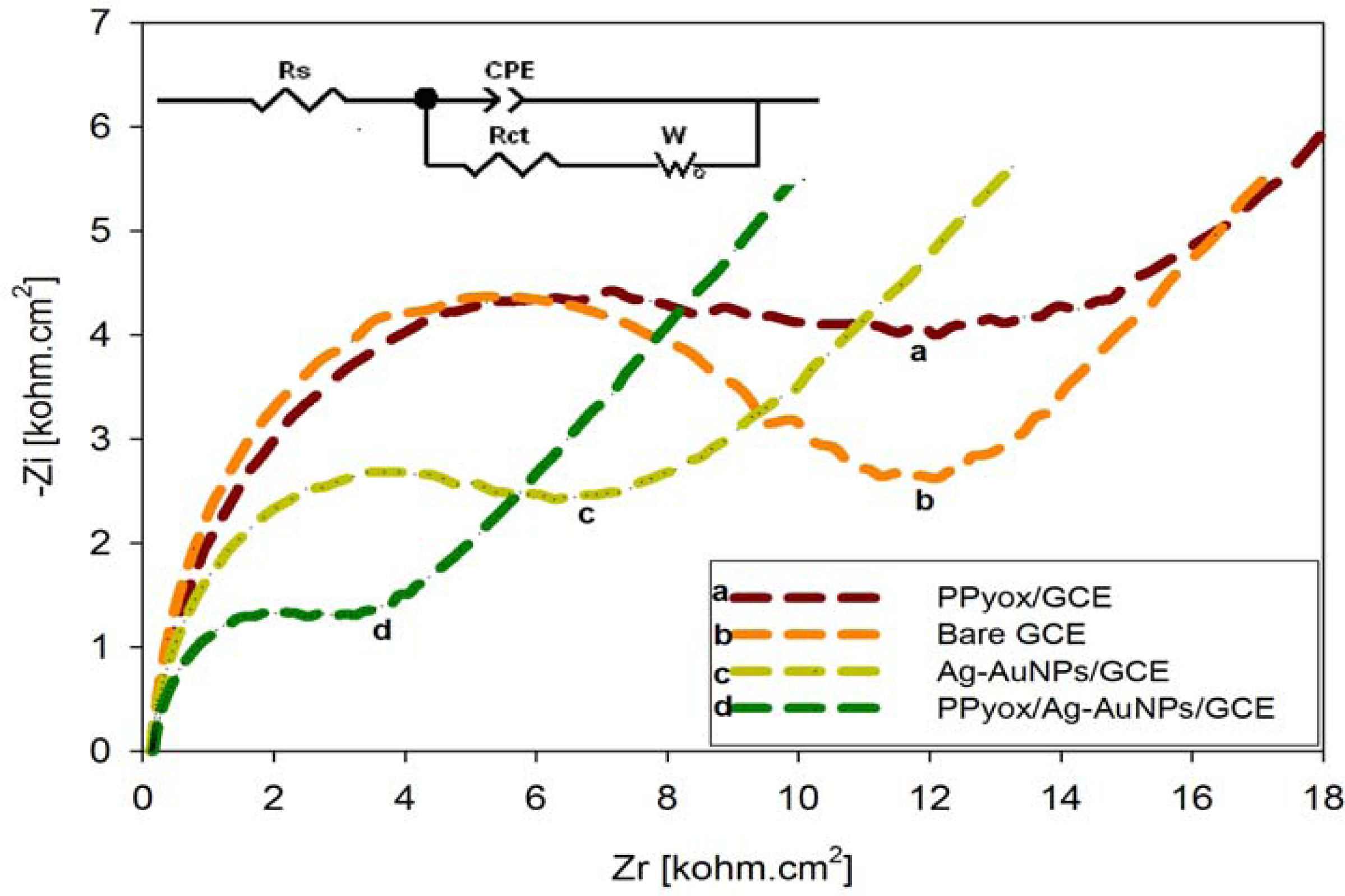
3.3.2. Electrochemical Impedance Spectroscopy (EIS) of the PPyox/Ag-AuNPs/GCE
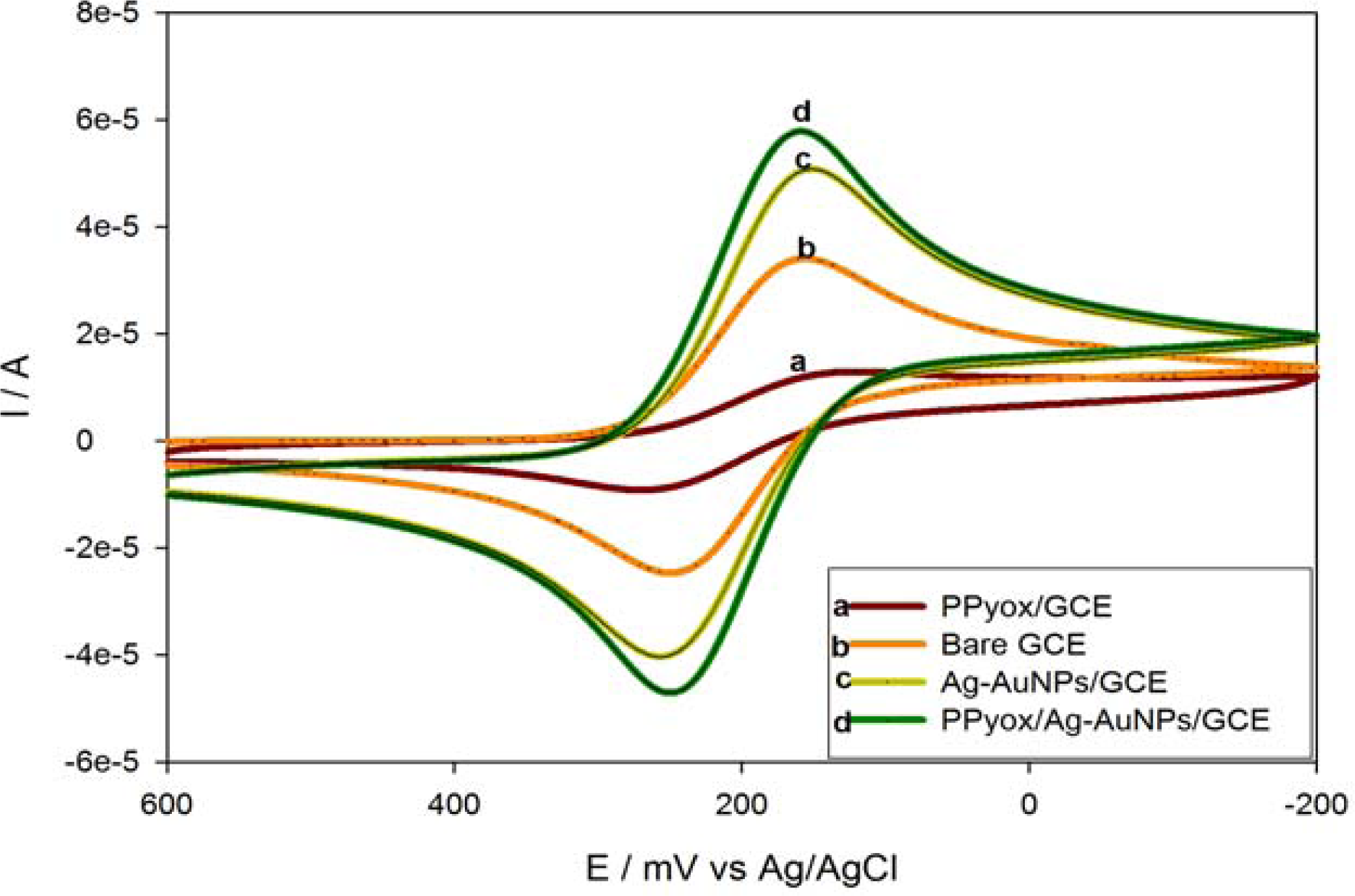
3.3.3. Cyclic Voltammetric Behaviour of the PPyox/Ag-AuNPs/GCE in the Presence of K3Fe(CN)6
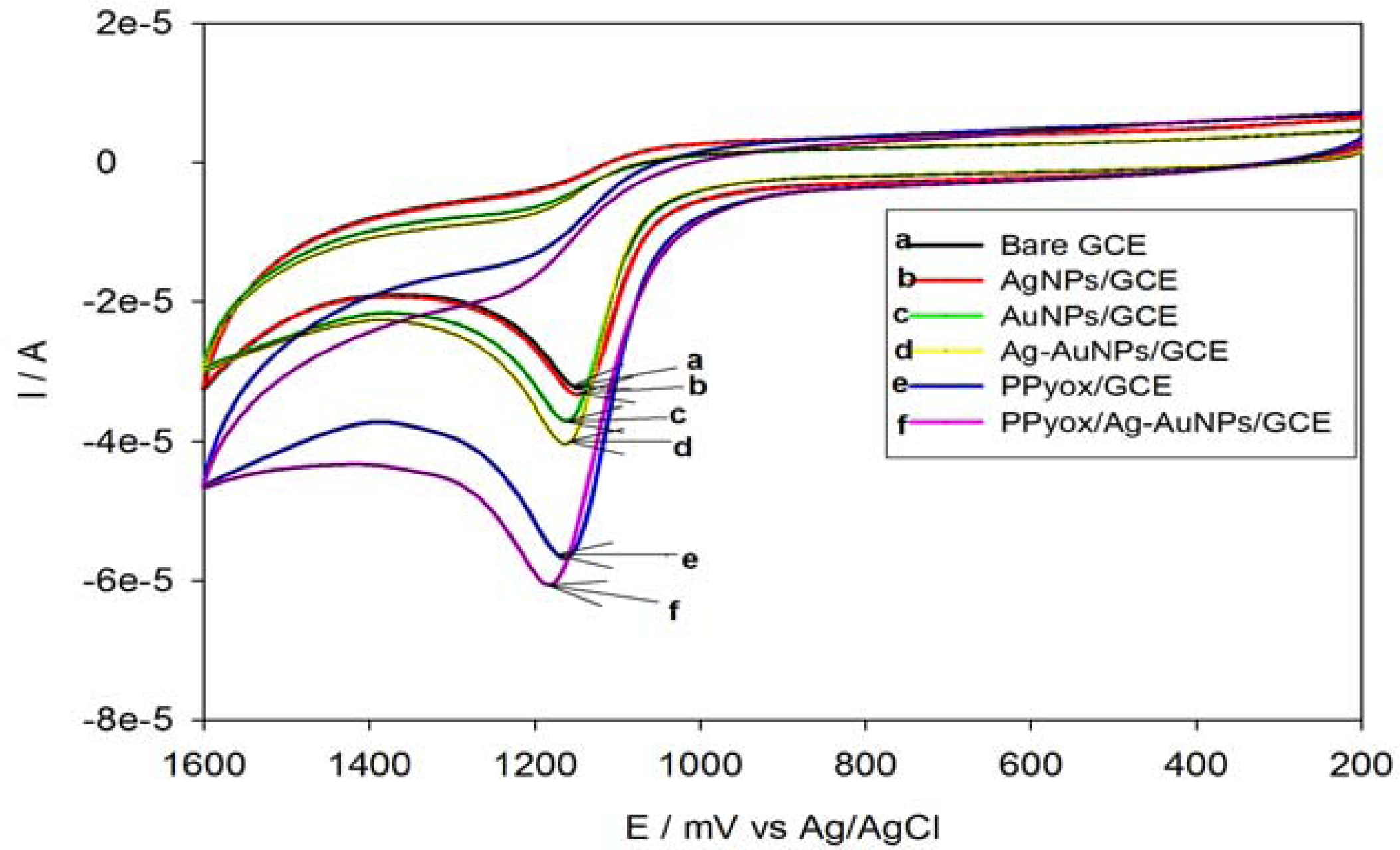
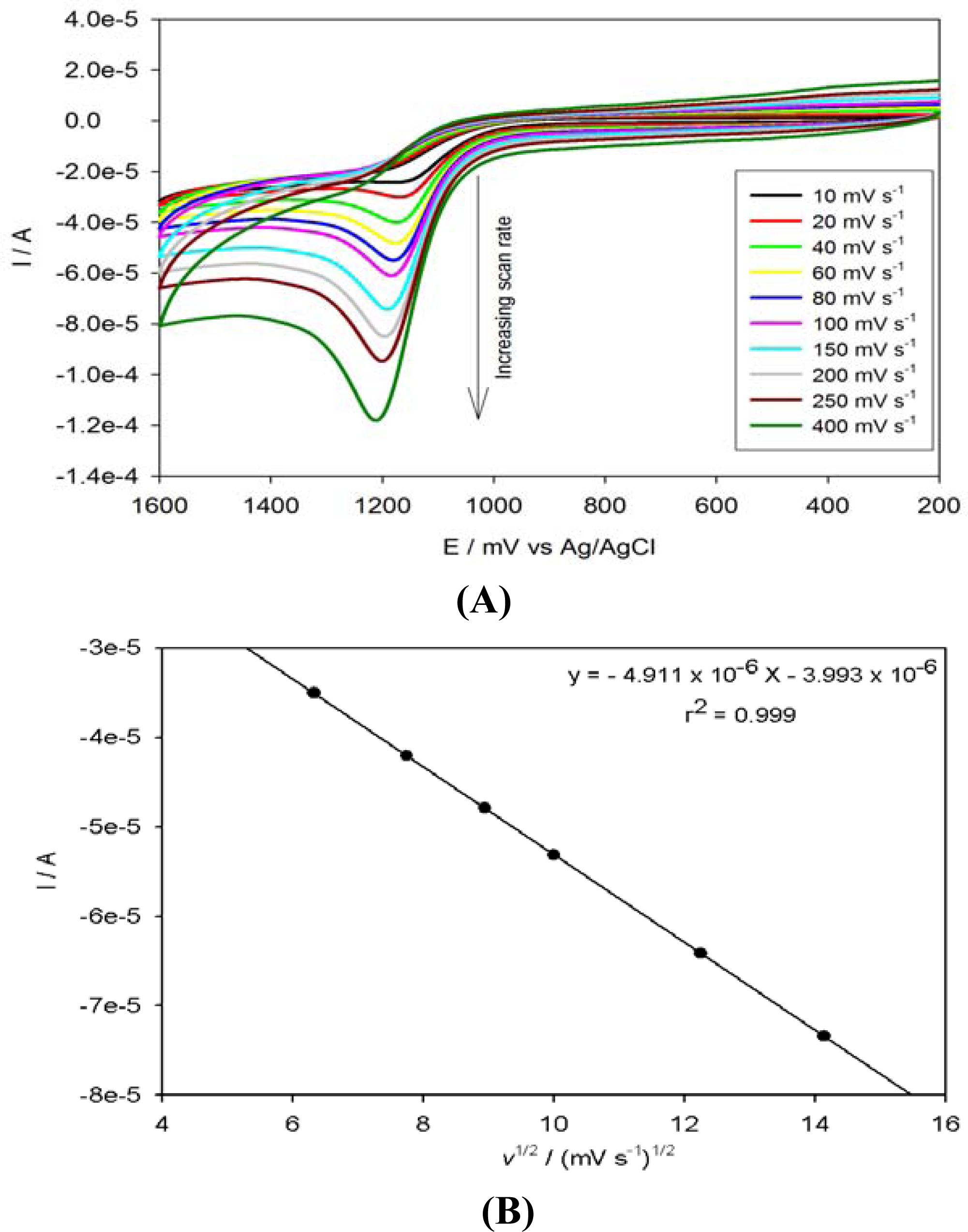
3.4. Electrocatalytic Oxidation of Anthracene on PPyox/Ag-AuNPs/GCE
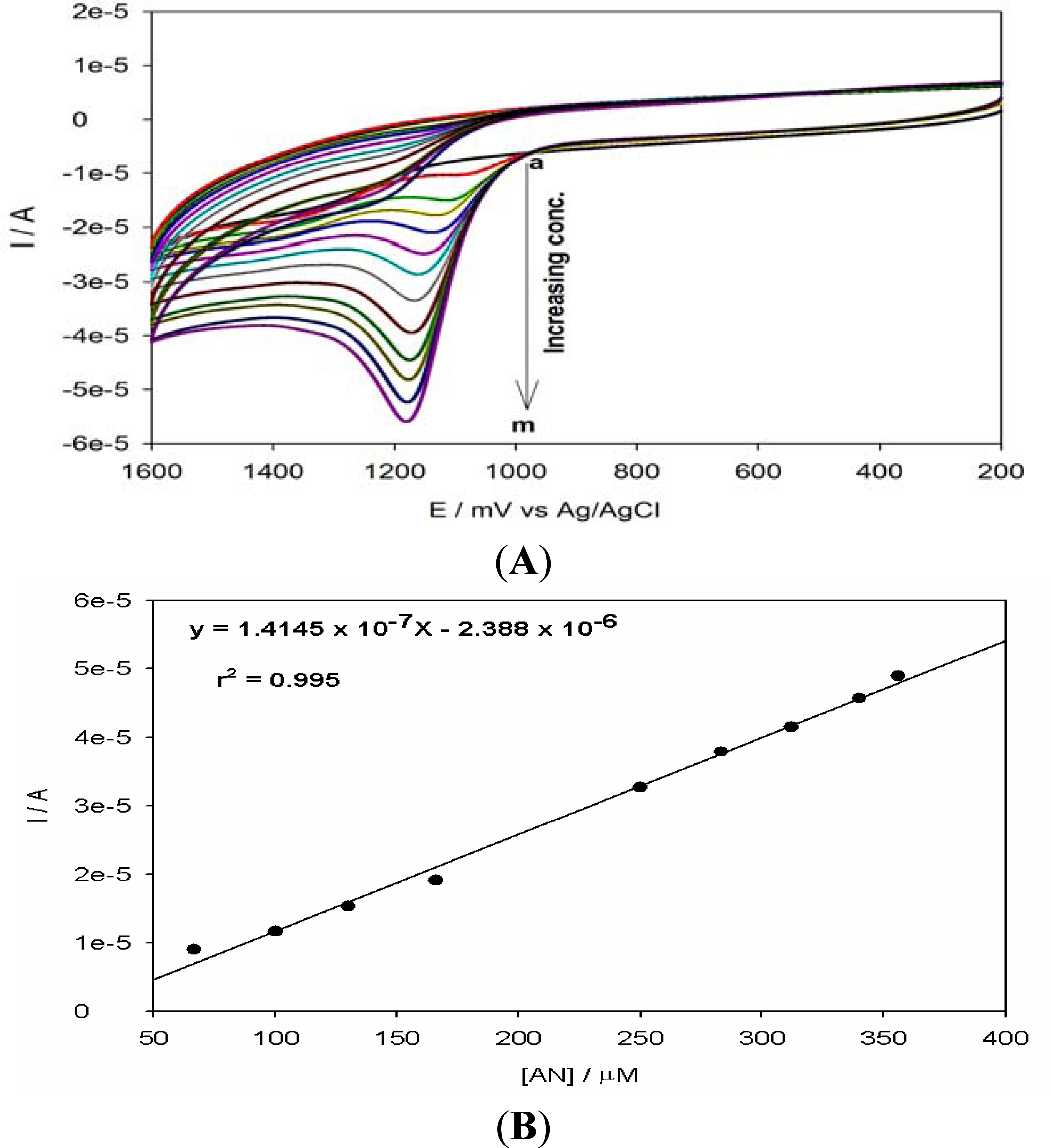
3.5. Analytical Application for Anthracene Detection
3.6. Reproducibility, Stability and Interference Studies
4. Conclusions
Acknowledgments
References
- Fetzer, JC. The Chemistry and Analysis of the Large Polyaromatic Hydrocarbons. Polyaromatic Hydrocarbons 2007, 27, 143–162. [Google Scholar]
- Beltrán, JL; Ferrer, R; Guiteras, M; Multivariate, J. Calibration of Polycyclic Aromatic Hydrocarbon Mixtures from Excitation-Emission Fluorescence Spectra. Anal. Chim. Acta 1998, 373, 311–319. [Google Scholar]
- Lewitzka, F. On-line and in situ Detection of Polycyclic Aromatic Hydrocarbons (PAH) on Aerosols via Thermodesorption and Laser-Induced Fluorescence Spectroscopy. J. Anal. Chem 2000, 366, 408–414. [Google Scholar]
- Lyons, G. Effects of Polyaromatic Hydrocarbons (PAHs), a Briefing to Highlight WWFs Work in Marine Conservation; National Environmental Technology Centre, AEA Technology: London, UK, August 1997. [Google Scholar]
- Woodward, KLA. Determination of Polyaroamatic Hydrocarbon Using Immunoassays. Sci. Total Envir 1984, 32, 103. [Google Scholar]
- Colmsjo, AHN. The Handbook of Environmental Chemistry Part 1, PAHs and Related Coumpounds; Springer-Verlag: New York, NY, USA, 1998; p. 55. [Google Scholar]
- Ferrer, R; Guiteras, J; Beltrán, JL. Development of Fast-Scanning Fluorescence Spectra as a Detection System for High-Performance Liquid Chromatography and Determination of Polycyclic Aromatic Hydrocarbons in Water Samples. J. Chromatogr. A 1997, 779, 123–130. [Google Scholar]
- Andrade Eiroa, EVB; López Mahía, P; Muniategui Lorenzo, S; Prada Rodríguez, D; Fernández Fernández, E. Determination of Polycyclic Aromatic Hydrocarbons (PAHs) in a Complex Mixture by Second-Derivative Constant-Energy Synchronous Spectrofluorimetry. Talanta 2000, 51, 677–684. [Google Scholar]
- Szolar, OHJ. Separation of PAHs by Capillary Electrophoresis (CE) Equipped with Laser-Induced Fluorescence. J. Mol. Rec 1996, 9, 515. [Google Scholar]
- Geffard, O; Geffard, A; His, E; Budzinski, H. Assessment of the Bioavailability and Toxicity of Sediment-Associated Polycyclic Aromatic Hydrocarbons and Heavy Metals Applied to Crassostrea Gigas Embryos and Larvae. Mar. Pollut. Bull 2003, 46, 481–490. [Google Scholar]
- Cai, Z-Q; Ya-Xian, Z; Yong, Z. Simultaneous Determination of Dissolved Anthracene and Pyrene in Aqueous Solution by Synchronous Fluorimetry. Spectrochim. Acta Pt A-Mol. Bio 2008, 69, 130–133. [Google Scholar]
- Rajeshwar, K. Environmental Electrochemistry Fundamentals and Applications in Pollution Abatement, 2nd ed; Academic Press: New York, NY, USA, 1997. [Google Scholar]
- Ballarin, B; Cassani, MC; Scavetta, E; Tonelli, D. Self-Assembled Gold Nanoparticles Modified ITO Electrodes: The Monolayer Binder Molecule Effect. Electrochim. Acta 2008, 53, 8034–8044. [Google Scholar]
- Gao, ZQ. Electrochemical-Behaviour of Dopamine and Ascorbic Acid at Overoxidized Polypyrrole (Dodecyl Sulfate) Film-Coated Electrodes. Anal. Chim. Acta 1993, 284, 393–404. [Google Scholar]
- Fang, J; Zhong, C; Mu, R. The Study of Deposited Silver Particulate Films by Simple Method for Efficient, SERS. Chem. Phys. Lett 2005, 401, 271–275. [Google Scholar]
- Asta Sileikaite, IP; Judita, P; Algimantas, J; Asta, G. Analysis of Silver Nanoparticles Produced by Chemical Reduction of Silver Salt Solution. Mater. Sci 2006, 12, 4. [Google Scholar]
- McFarland, AD; Mirkin, CA; Van Duyne, RP; Godwin, HA. Synthesis of Fold Nanoparticles. J. Chem. Educ 2004, 81, 544. [Google Scholar]
- Pal, A; Shah, S; Kulkarni, V; Murthy, RSR; Devi, S. Template Free Synthesis of Silver-Gold Alloy Nanoparticles and Cellular Uptake of Gold Nanoparticles in Chinese Hamster Ovary Cell. Mater. Chem. Phys 2009, 113, 276–282. [Google Scholar]
- Shin, Y; Bae, IT; Bruce, WA; Gregory, JE. Facile Stabilization of Gold-Silver Alloy Nanoparticles on Cellulose Nanocrystal. J. Phys. Chem 2008, 112, 4844–4848. [Google Scholar]
- Zhu, J. Theoretical Study of the Optical Absorption Properties of Au-Ag Bimetallic Nanospheres. Phys. E: Low-Dimens. Syst. Nanostr 2005, 27, 296–301. [Google Scholar]
- Angshuman, PA. Preparation of Silver—Gold Alloy Nanoparticles at Higher Concentration Using Sodium Dodecyl Sulfate. J. Chem 2008, 61, 66. [Google Scholar]
- Link, S; El-Sayed, MA. Alloy Formation of Gold—Silver Nanoparticles and the Dependence of the Plasmon Absorption on Their Composition. J. Phys. Chem 1999, 103, 3529–3533. [Google Scholar]
- Alqudami, ASA; Shivaprasad, GSM. Ag-Au Alloy Nanoparticles Prepared by Electro-Exploding Wire Technique. J. Nanopart. Res 2007, 10, 1027–1036. [Google Scholar]
- Fernàndez-Sańchez, C; McNeil, CJ; Rawson, K. Electrochemical Impedance Spectroscopy Studies of Polymer Degradation: Application to Biosensor Development. Trends Anal. Chem 2005, 24, 37–48. [Google Scholar]
- Ehret, R; Baumann, W; Brischwein, M; Schwinde, A; Stegbauer, K; Wolf, B. Monitoring of Cellular Behaviour by Impedance Measurements on Interdigitated Electrode Structures. Biosens. Bioelectron 1997, 12, 29–41. [Google Scholar]
- Kang, X; Mai, Z; Zou, X; Cai, P; Mo, J. A Novel Glucose Biosensor Based on Immobilization of Glucose Oxidase in Chitosan on a Glassy Carbon Electrode Modified with Gold-Platinum Alloy Nanoparticles/Multiwall Carbon Nanotubes. Anal. Biochem 2007, 369, 71–79. [Google Scholar]
- Beck, F; Oberst, M; Bunsenges, B. Organic Electrochemistry in the Solid State-Overoxidation of Polypyrole. J. Phys. Chem 1987, 91, 967–974. [Google Scholar]
- Christensen, PA; Hamnett, A. In situ Spectroscopic Investigations of the Growth, Electrochemical Cycling and Overoxidation of Polypyrrole in Aqueous Solution. Electrochim. Acta 1991, 36, 1263–1286. [Google Scholar]
- Martin, CR; Van Dyke, LS; Cai, Z. Template-Synthesis—A Method for Enhancing the Ionic and Electronic Conductivity in Electronically Conductive Polymers. Electrochim. Acta 1992, 37, 1611–1613. [Google Scholar]
- van Dyke, LS; Martin, CR. Fibrillar Electronically Conductive Polymers Show Enhanced Rates of Charge Transport. Syn. Metals 1990, 36, 275–281. [Google Scholar]
- Li, J; Lin, X-Q. Electrodeposition of Gold Nanoclusters on Overoxidized Polypyrrole Film Modified Glassy Carbon Electrode and its Application for the Simultaneous Determination of Epinephrine and Uric Acid Under Coexistence of Ascorbic Acid. Anal. Chim. Acta 2007, 596, 222–230. [Google Scholar]
- Tominaga, M; Shimazoe, T; Nagashima, M; Taniguchi, I. Composition-Activity Relationships of Carbon Electrode-Supported Bimetallic Gold-Silver Nanoparticles in Electrocatalytic Oxidation of Glucose. J. Electroanal. Chem 2008, 615, 51–61. [Google Scholar]
- Mathebe, NGR; Morrin, A; Iwuoha, EI. Electrochemistry and Scanning Electron Microscopy of Polyaniline/Peroxidase-Based Biosensor. Talanta 2004, 64, 115–120. [Google Scholar]
- Bouvrette, P; Hrapovic, S; Male, KB; Luong, JHT. Analysis of the 16 Environmental Protection Agency Priority Polycyclic Aromatic Hydrocarbons by High Performance Liquid Chromatography-Oxidized Diamond Film Electrodes. J.Chromatogr. A 2006, 1103, 248–256. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Epa (mV) | Epc (mV) | Ipa (A) | Ipc (A) | ΔEp | Eo' |
|---|---|---|---|---|---|---|
| PPyox/GCE | 264 | 125 | −1.189 × 10−5 | 1.170 × 10−5 | 143 | 194.5 |
| Bare GCE | 248 | 157 | −3.328 × 10−5 | 3.357 × 10−5 | 91 | 202.5 |
| Ag-AuNPs/GCE | 255 | 152 | −5.103 × 10−5 | 5.186 × 10−5 | 103 | 203.5 |
| PPyox/Ag-AuNPs/GCE | 249 | 159 | −5.919 × 10−5 | 5.939 × 10−5 | 90 | 204.0 |
| Electrode | Epa (mV) | Ipa (A) |
|---|---|---|
| Bare GCE | 1,148 | 2.778 × 10−5 |
| AgNps/GCE | 1,150 | 2.900 × 10−5 |
| AuNPs/GCE | 1,160 | 3.193 × 10−5 |
| Ag-AuNPs/GCE | 1,162 | 3.537 × 10−5 |
| PPyox/GCE | 1,162 | 4.657 × 10−5 |
| PPyox/Ag-AuNPs/GCE | 1,181 | 5.225 × 10−5 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mailu, S.N.; Waryo, T.T.; Ndangili, P.M.; Ngece, F.R.; Baleg, A.A.; Baker, P.G.; Iwuoha, E.I. Determination of Anthracene on Ag-Au Alloy Nanoparticles/Overoxidized-Polypyrrole Composite Modified Glassy Carbon Electrodes. Sensors 2010, 10, 9449-9465. https://doi.org/10.3390/s101009449
Mailu SN, Waryo TT, Ndangili PM, Ngece FR, Baleg AA, Baker PG, Iwuoha EI. Determination of Anthracene on Ag-Au Alloy Nanoparticles/Overoxidized-Polypyrrole Composite Modified Glassy Carbon Electrodes. Sensors. 2010; 10(10):9449-9465. https://doi.org/10.3390/s101009449
Chicago/Turabian StyleMailu, Stephen N., Tesfaye T. Waryo, Peter M. Ndangili, Fanelwa R. Ngece, Abd A. Baleg, Priscilla G. Baker, and Emmanuel I. Iwuoha. 2010. "Determination of Anthracene on Ag-Au Alloy Nanoparticles/Overoxidized-Polypyrrole Composite Modified Glassy Carbon Electrodes" Sensors 10, no. 10: 9449-9465. https://doi.org/10.3390/s101009449