A Nitrite Biosensor Based on Co-immobilization of Nitrite Reductase and Viologen-modified Chitosan on a Glassy Carbon Electrode

Abstract
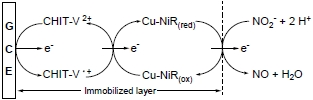
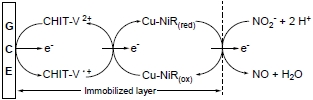
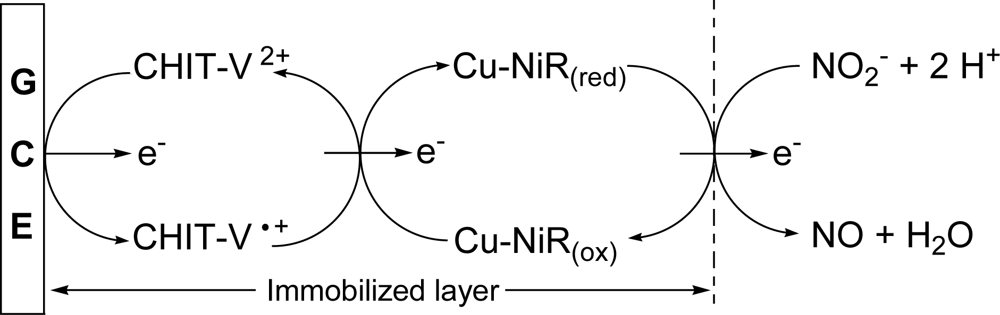
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Chemicals and Materials
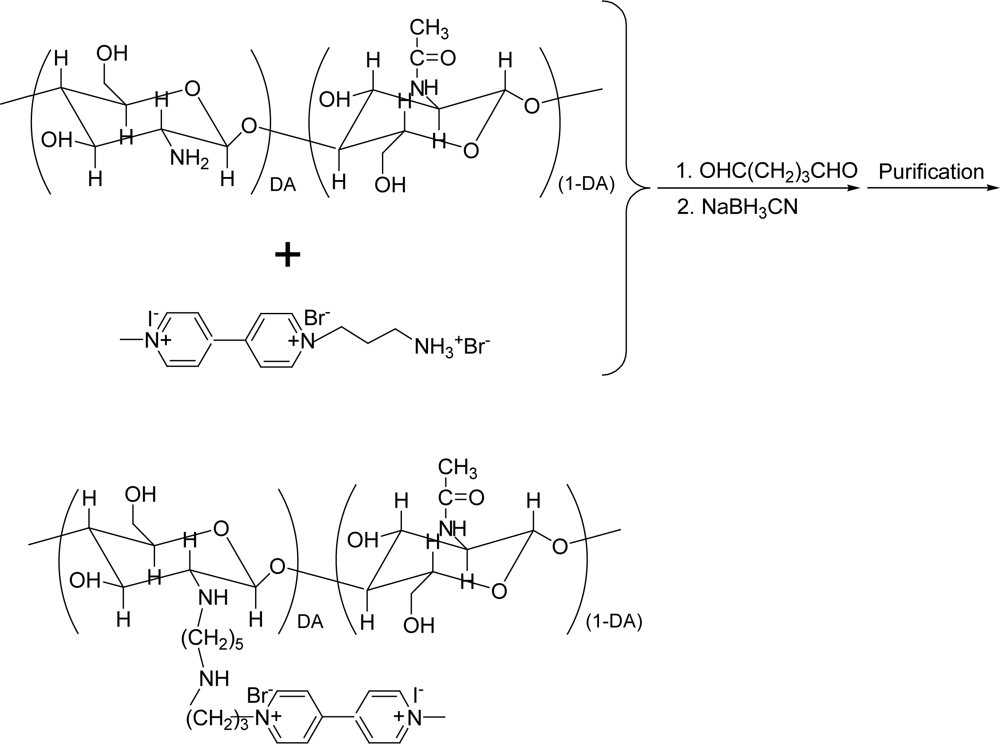
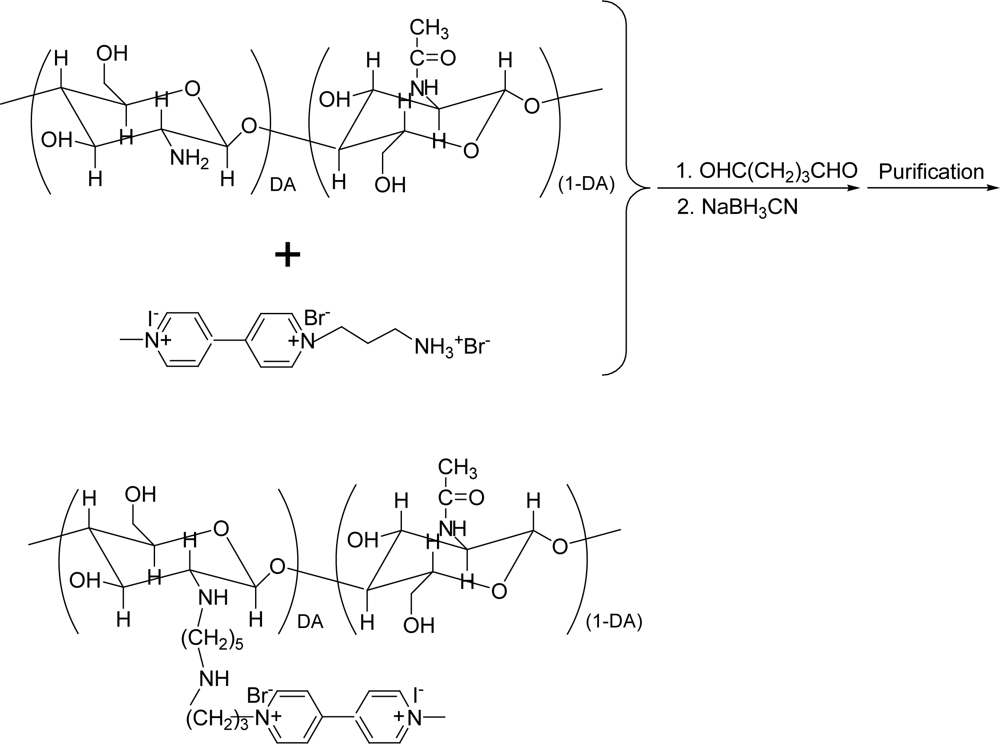
2.2. Synthesis of CHIT-V
2.3. Co-immobilization of NiR and CHIT-V
2.4. Apparatus
3. Results and Discussion
3.1. Synthesis of CHIT-V
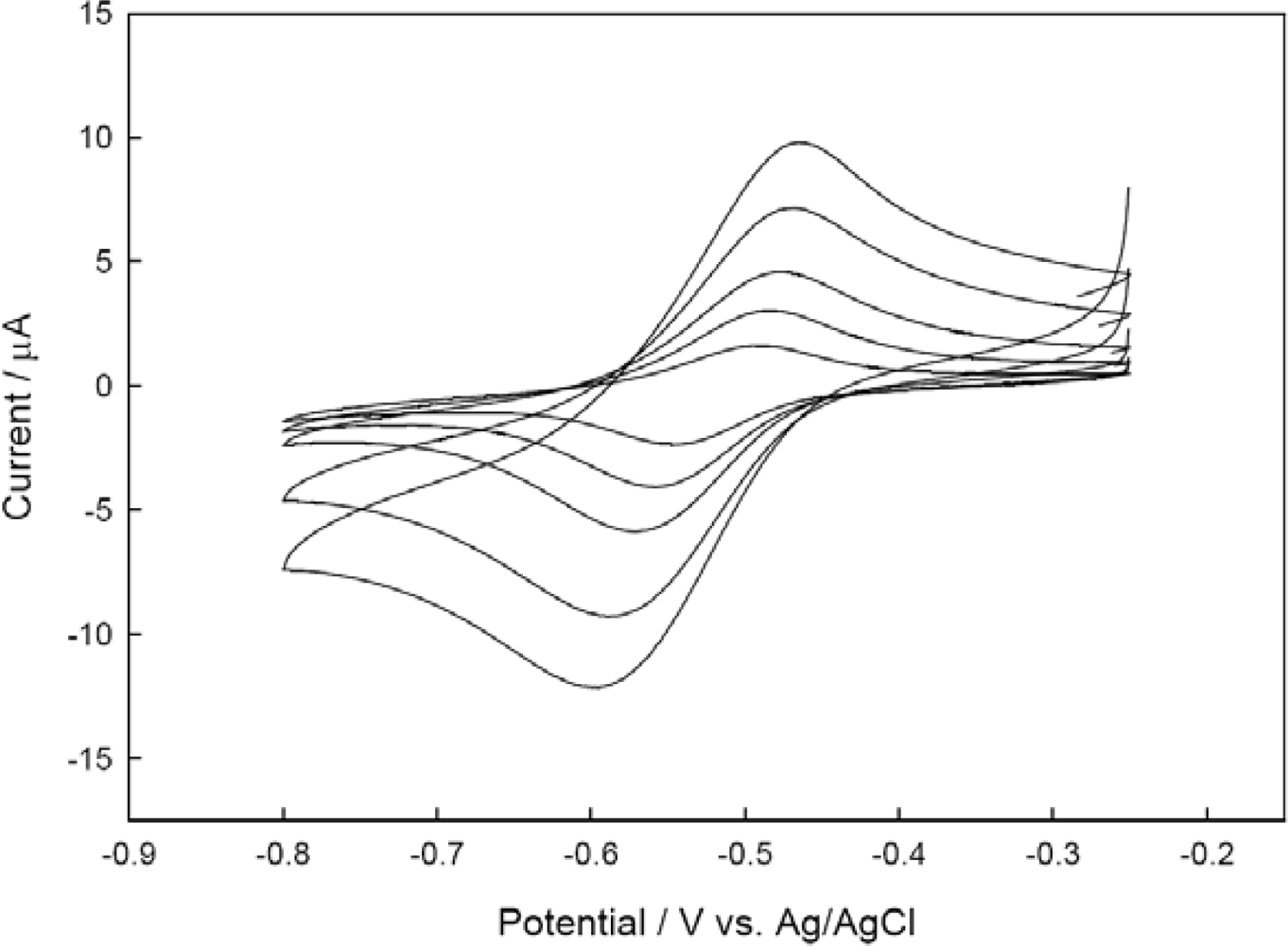
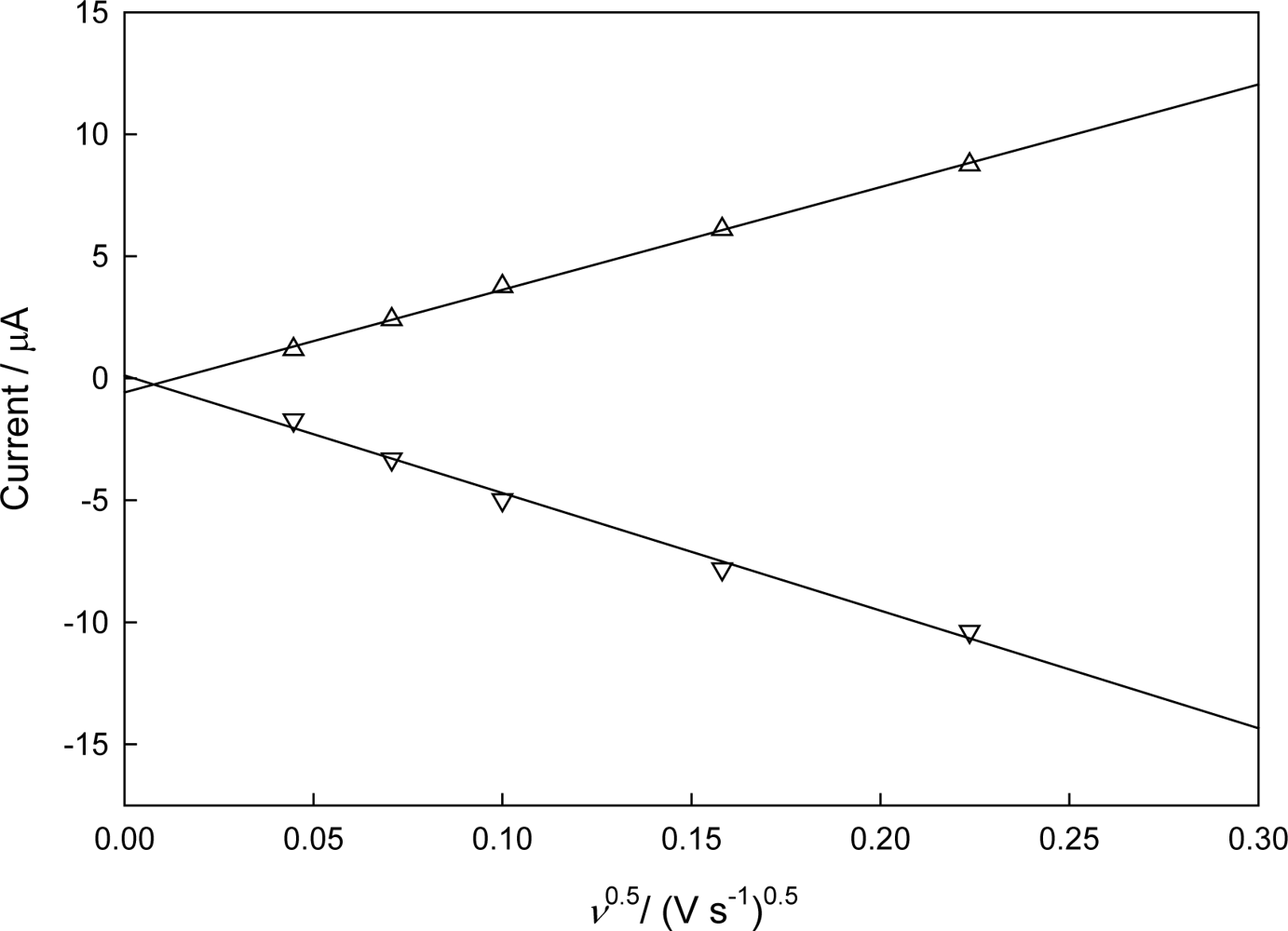
3.2. Electrochemical Characteristics of Immobilized CHIT-V
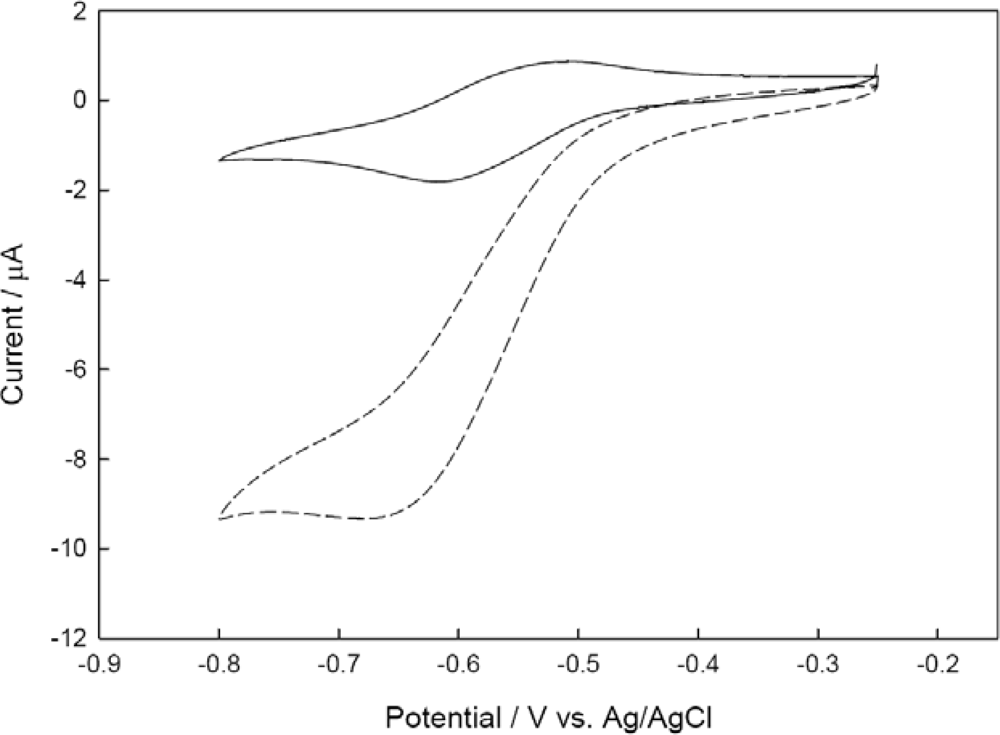
3.3. Catalytic Behavior of the Co-immobilized NiR and CHIT-V GCE
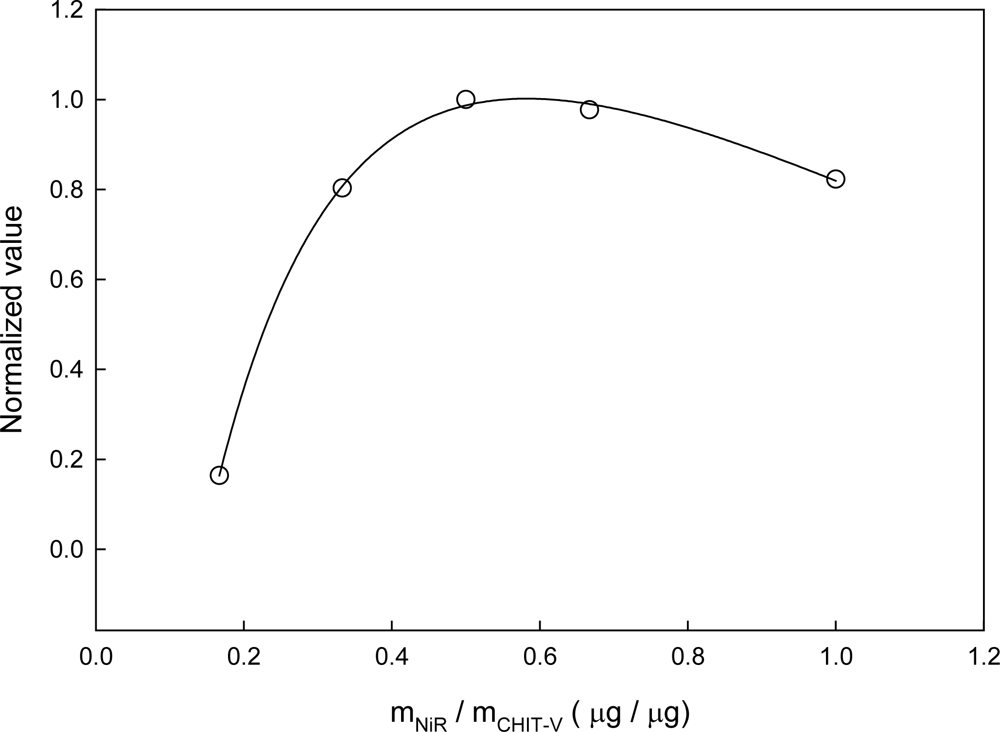
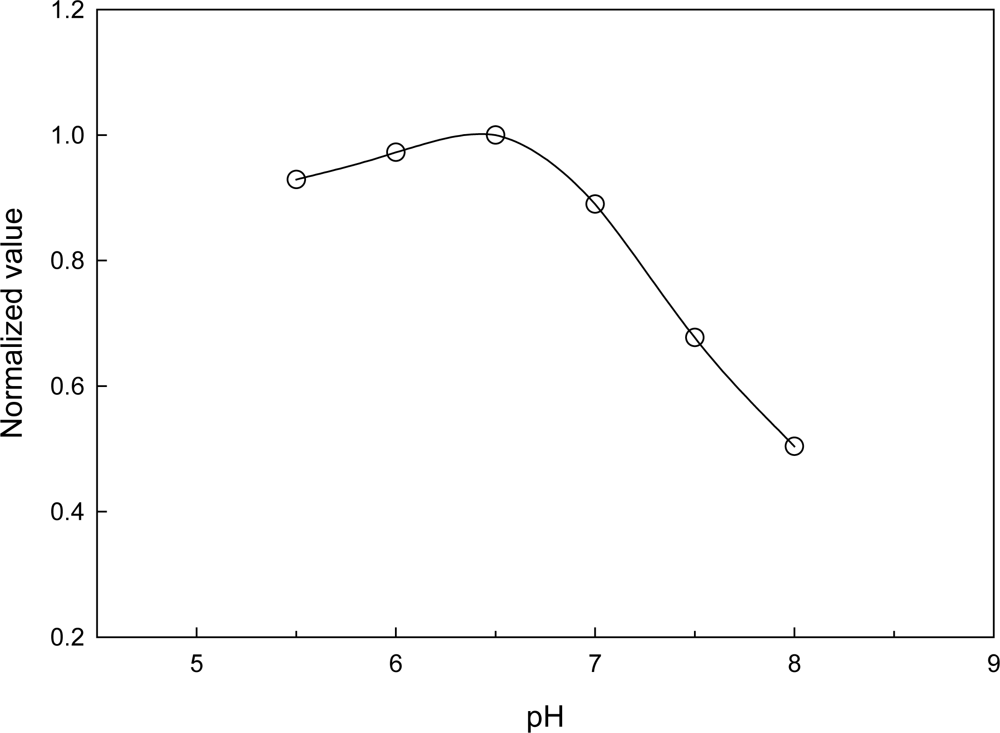
3.4. Optimization of the Co-immobilized NiR and CHIT-V GCE
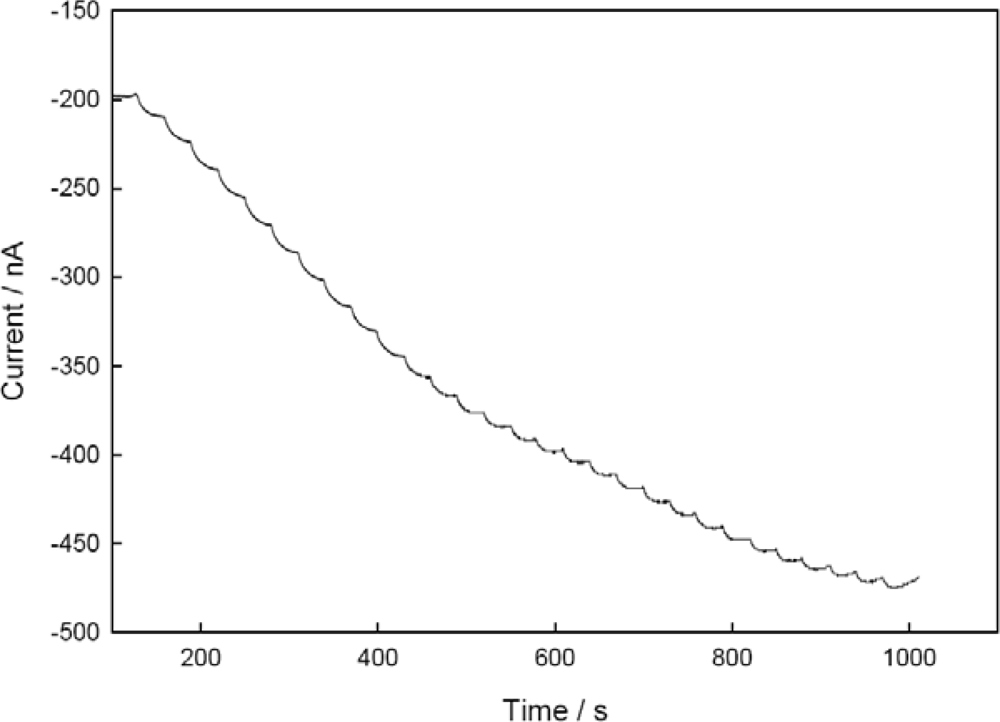
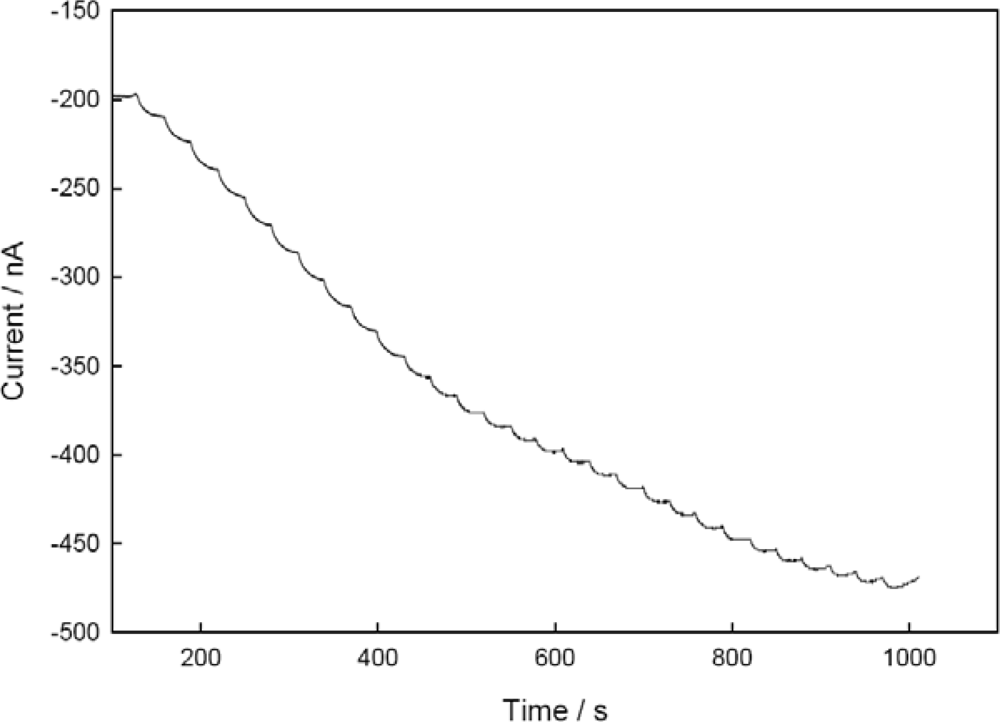
3.5. Performance Factors of Co-immobilized NiR and CHIT-V on GCE as a Biosensor
4. Conclusions
Acknowledgments
References
- Kiang, C.H.; Kuan, S.S.; Guibault, G.G. Enzymatic determination of nitrate: electrochemical detection after reduction with nitrate reductase and nitrite reductase. Anal. Chem 1978, 50, 1319–1322. [Google Scholar]
- Rajeshwar, K.; Ibanez, J.G. Environmental Electrochemistry; Academic Press: Durham, NC, USA, 1997; p. 29. [Google Scholar]
- Moorcroft, M.J.; Davis, J.; Compton, R.G. Detection and determination of nitrate and nitrite: a review. Talanta 2001, 54, 785–803. [Google Scholar]
- McCarthy, P.; Klusman, R.W.; Cowling, S.W.; Rice, J.A. Water analysis. Anal. Chem 1993, 65, 244R–292R. [Google Scholar]
- Clesceri, L.S.; Greenberg, A.E.; Eaton, A.D. Standard Methods for the Examination of Water and Wastewater, 20th Ed ed; American Public Health Association: Washington, DC, USA, 1998; part 4000. [Google Scholar]
- Hamano, T.; Mitsuhashi, Y.; Aoki, N.; Semma, M.; Ito, Y.; Oji, Y. Enzymatic method for the determination of nitrite in meat and fish products. Analyst 1998, 123, 1127–1129. [Google Scholar]
- Ferretti, S.; Lee, S-K.; MacCraith, B.D.; Oliva, A.G.; Richardson, D.J.; Russell, D.A.; Sapsford, K.E.; Vidal, M. Optical biosensing of nitrite ions using cytochrome cd1 nitrite reductase encapsulated in a sol-gel matrix. Analyst 2000, 125, 1993–1999. [Google Scholar]
- Rosa, C.C.; Cruz, H.J.; Vidal, M.; Oliva, A.G. Optical biosensor based on nitrite reductase immobilized in controlled pore glass. Biosens. Bioelectron 2002, 17, 45–52. [Google Scholar]
- Zhang, Z.; Xia, S.; Leonard, D.; Jaffrezic-Renault, N.; Zhang, J.; Bessueille, F.; Goepfert, Y.; Wang, X.; Chen, L.; Zhu, Z.; Zhao, J.; Almeida, M.G.; Silveira, C.M. A novel nitrite biosensor based on conductrometric electrode modified with cytochrome c nitrite reductase composite membrane. Biosens. Bioelectron 2009, 24, 1574–1579. [Google Scholar]
- Kiang, C.H.; Kuan, S.S.; Guibault, G.G. A novel enzyme electrode method for the determination of nitrite based on nitrite reductase. Anal. Chim. Acta 1975, 80, 209–214. [Google Scholar]
- Scharf, M.; Moreno, C.; Costa, C.; van Dijk, C.; Payne, W.P.; LeGall, J.; Moura, I.; Moura, J.J.G. Electrochemical studies on nitrite reductase towards a biosensor. Biochem. Biophys. Res. Commun 1995, 209, 1018–1025. [Google Scholar]
- Strehlitz, B.; Gründig, B.; Schumacher, W.; Kronack, P.M.H.; Vorlop, K.-D.; Kotte, H. A nitrite sensor based on a highly sensitive nitrite reductase mediator-coupled amperometric detection. Anal. Chem 1996, 68, 807–816. [Google Scholar]
- Wu, Q.; Storrier, G.D.; Pariente, F.; Yang, Y.; Shapleigh, J.P.; Abruňa, H.D. A nitrite biosensor based on a maltose binding protein nitrite reductase fusion immobilized on an electropolymerized film of a pyrrole-derived bipyridinium. Anal. Chem 1997, 69, 4856–4863. [Google Scholar]
- Sasaki, S.; Karube, I.; Hirota, N.; Arikawa, Y.; Nishiyama, M.; Kukimoto, M.; Horinouchi, S.; Beppu, T. Application of nitrite reductase from Alcaligenes faecalis S-6 for nitrite measurement. Biosens. Bioelectron 1998, 13, 1–5. [Google Scholar]
- Silva, S.D.; Cosnier, S.; Almeida, M.G.; Moura, J.J.G. An efficient poly(pyrrole-viologen)-nitrite reductase biosensor for the mediated detection of nitrite. Electrochem. Commun 2004, 6, 404–408. [Google Scholar]
- Astier, Y.; Canters, G.W.; Davis, J.J.; Hill, H.A.O.; Verbeet, M.P.; Wijma, H.J. Sensing nitrite through a pseudoazurin-nitrite reductase electron transfer relay. Chemphyschem 2005, 6, 1114–1120. [Google Scholar]
- Quan, D.; Min, D.G.; Cha, G.S.; Nam, H. Electrochemical characterization of biosensor based on nitrite reductase and methyl viologen co-immobilized glassy carbon electrode. Bioelectrochemistry 2006, 69, 267–275. [Google Scholar]
- Almeida, M.G.; Silveira, C.M.; Moura, J.J.G. Biosensing nitrite using the system nitrite reductase/Nafion/methyl viologen: A voltammetric study. Biosens. Bioelectron 2007, 22, 2485–2492. [Google Scholar]
- Chen, H.; Mousty, C.; Cosnier, S.; Silveira, C.; Moura, J.J.G.; Almeida, M.G. Highly sensitive nitrite biosensor based on the electrical wiring of nitrite reductase by [ZnCr-AQS] LDH. Electrochem. Commun 2007, 9, 2240–2245. [Google Scholar]
- Kozuma, T.; Shidara, S.; Yamaguchi, K.; Nakamura, N.; Deligeer; Suzuki, S. Direct electrochemistry of copper-containing nitrite reductase from Achromobacter xylosoxidans NCIB 11015. Chem. Lett 1993, 12, 2029–2032. [Google Scholar]
- Kozuma, T.; Shidara, S.; Suzuki, S. Direct electrochemistry of nitrite reductase from Achromobacter cycloclastes IAM 1013. Bull. Chem. Soc. Jpn 1994, 67, 138. [Google Scholar]
- Quan, D.; Nagarale, R.K.; Shin, W. A nitrite biosensor based on co-immobilization of nitrite reductase and viologen-modified polysiloxane on glassy carbon electrode. Electroanalysis 2010, in press.. [Google Scholar]
- Godden, J.W.; Turley, S.; Teller, D.C.; Adman, E.T.; Liu, M.; Payne, W.J.; JeGall, J. The 2.3 angstrom X-ray structure of nitrite reductase from Achromobacter cycloclastes. Science 1991, 253, 438–442. [Google Scholar]
- Kukimoto, M.; Nishiyama, M.; Murphy, M.E.P.; Turley, S.; Adman, E.T.; Horinouchi, S.; Beppu, T. X-ray structure and site-directed mutagenesis of a nitrite reductase from Acaligenes faecalis S-6: roles of two copper atoms in nitrite reduction. Biochemistry 1994, 33, 5246–5252. [Google Scholar]
- Adman, E.T.; Godden, J.W.; Turley, S. The structure of copper-nitrite reductase from Achromobacter cycloclastes at five pH values, with NO2− bound and with type II copper depleted. J. Biol. Chem 1995, 270, 27458–27474. [Google Scholar]
- Averill, B.A. Dissimilatory nitrite and nitric oxide reductase. Chem. Rev 1996, 96, 2951–2964. [Google Scholar]
- Murphy, M.E.P.; Turley, S.; Adman, E.T. Structure of nitrite bound to copper-containing nitrite reductase from Acaligenes faecalis. J. Biol. Chem 1997, 272, 28455–28460. [Google Scholar]
- Olesen, K.; Veselov, A.; Zhao, Y.; Wang, Y.; Danner, B.; Scholes, C.P.; Shapleigh, J.P. Spectroscopic, kinetic, and electrochemical characterization of heterologously expressed wild-type and mutant forms of copper-containing nitrite reductase from Rhodobacter sphaeroides 2.4.3. Biochemistry 1998, 37, 6086–6094. [Google Scholar]
- Cutruzzolá, F. Baterial nitric oxide synthesis. Biochim. Biophys. Acta 1999, 1411, 231–249. [Google Scholar]
- Suzuki, S.; Kataoka, K.; Yamaguchi, K. Metal coordination and mechanism of multicopper nitrite reductase. Acc. Chem. Res 2000, 33, 728–735. [Google Scholar]
- Zhao, Y.; Likoyanov, D.A.; Toropov, Y.V.; Wu, K.; Shapleigh, J.P.; Scholes, C.P. Catalytic function and local proton structure at the type 2 copper of nitrite reductase: the correlation of enzymatic pH dependence, conserved residues, and proton hyperfine structure. Biochemistry 2002, 41, 7464–7474. [Google Scholar]
- Ellis, M.J.; Dodd, F.E.; Sawers, G.; Eady, R.R.; Hasnain, S.S. Atomic resolution structures of native copper nitrite reductase from Alcaligenes xylosoxidans and the active site mutant Asp92Glu. J. Mol. Biol 2003, 328, 429–438. [Google Scholar]
- Kataoka, K.; Yamaguchi, K.; Kobayashi, M.; Mori, T.; Bokui, N.; Suzuki, S. Structure-based engineering of Alcaligenes xylosoxidans copper-containing nitrite reductase enhances intermolecular electron transfer reaction with pseudoazurin. J. Biol. Chem 2004, 279, 53374–53378. [Google Scholar]
- Yokoyama, H.; Yamaguchi, K.; Sugimoto, M.; Suzuki, S. CuI and CuII complexes containing nitrite and tridentate aromatic amine ligand as models for the substrate-binding type-2 Cu site of nitrite reductase. Eur. J. Inorg. Chem 2005, 2005, 1435–1441. [Google Scholar]
- Tocheva, E.I.; Eltis, L.D.; Murphy, M.E. Conserved active site residues limit inhibition of a copper-containing nitrite reductase by small molecules. Biochemistry 2008, 47, 4452–4460. [Google Scholar]
- Michalski, W.P.; Nicholas, D.J.D. Molecular characterization of a copper-containing nitrite reductase from Rhodopseudomonas sphaeroides forma sp. denitrificans. Biochim. Biophys. Acta 1985, 828, 130–137. [Google Scholar]
- Stripe, A.; Guzzi, R.; Wijma, H.; Verbeet, M.Ph.; Canters, G.W.; Sportelli, L. Calorimetric and spectroscopic investigations of the thermal denaturation of wild type nitrite reductase. Biochim. Biophys. Acta 2005, 1752, 47–55. [Google Scholar]
- Pinho, D.; Besson, S.; Brondino, C.D.; de Castro, B.; Moura, I. Copper-containing nitrite reductase from Pseudomanas chlororaphis DSM 50135. Eur. J. Biochem 2004, 271, 2361–2369. [Google Scholar]
- Yi, H.; Wu, L.; Bentley, W.E.; Gohdssi, R.; Rubloff, G.W.; Culver, J.N.; Payne, G.F. Biofabrication with chitosan: a review. Biomacromolecules 2005, 6, 2881–2894. [Google Scholar]
- Macquarrie, D.J.; Hardy, J.J.E. Applications of functionalized chitosan in catalysis. Ind. Eng. Chem. Res 2005, 44, 8499–8520. [Google Scholar]
- Krajewska, B. Application of chitin- and chitosan-based materials for enzyme immobilizations: a review. Enzyme Microb. Tech 2004, 35, 126–139. [Google Scholar]
- Krajewska, B. Membrane-based processes performed with use of chitin/chitosan materials: a review. Sep. Purif. Tech 2005, 41, 305–312. [Google Scholar]
- Vazquez-Duhalt, R.; Tinoco, R.; Topoleski, P.; D’Antonio, L.D.; Payne, G.F. Enzyme conjugation to the polysaccharide chitosan: smart biocatalysts and biocatalytic hydrogels. Bioconjugate Chem 2001, 12, 301–306. [Google Scholar]
- Okeke, B.C.; Ma, G.; Cheng, Q.; Losi, M.E.; Frankenberger, W.T., Jr. Development of perchlorate reductase-based biosensor for real time analysis of perchlorate in water. J. Microbiol. Meth 2007, 68, 69–75. [Google Scholar]
- Zhang, M.; Gorski, W. Electrochemical sensing platform based on the carbon nanotubes/redox mediators-biopolymer system. J. Am. Chem. Soc 2005, 127, 2058–2059. [Google Scholar]
- Tangkuaram, T.; Wang, J.; Rodriguez, M.C.; Laocharoensuk, R.; veerasai, W. Highly stable amplified low-potential electrocatalytic detection of NAD+ at azure-chitosan modified carbon electrodes. Sens. Actuat. B 2007, 121, 277–281. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.A.; Gurny, R. Structure and interactions in covently and ionically crosslinked chitosan hydrogels for biomedical applications: a review. Eur. J. Pharm. Biopharm 2004, 57, 19–34. [Google Scholar]
- Ferreyra, N.F.; Dassie, S.A.; Solis, V.M. Electroreduction of methyl viologen in the presence of nitrite. Its influence on enzymatic electrodes. J. Electroanal. Chem 2000, 486, 126–132. [Google Scholar]







© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quan, D.; Shin, W. A Nitrite Biosensor Based on Co-immobilization of Nitrite Reductase and Viologen-modified Chitosan on a Glassy Carbon Electrode. Sensors 2010, 10, 6241-6256. https://doi.org/10.3390/s100606241
Quan D, Shin W. A Nitrite Biosensor Based on Co-immobilization of Nitrite Reductase and Viologen-modified Chitosan on a Glassy Carbon Electrode. Sensors. 2010; 10(6):6241-6256. https://doi.org/10.3390/s100606241
Chicago/Turabian StyleQuan, De, and Woonsup Shin. 2010. "A Nitrite Biosensor Based on Co-immobilization of Nitrite Reductase and Viologen-modified Chitosan on a Glassy Carbon Electrode" Sensors 10, no. 6: 6241-6256. https://doi.org/10.3390/s100606241