Enzyme Immobilization Strategies and Electropolymerization Conditions to Control Sensitivity and Selectivity Parameters of a Polymer-Enzyme Composite Glucose Biosensor

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals and Solutions
2.2. Instrumentation and Software
2.3. Working Electrode Preparation
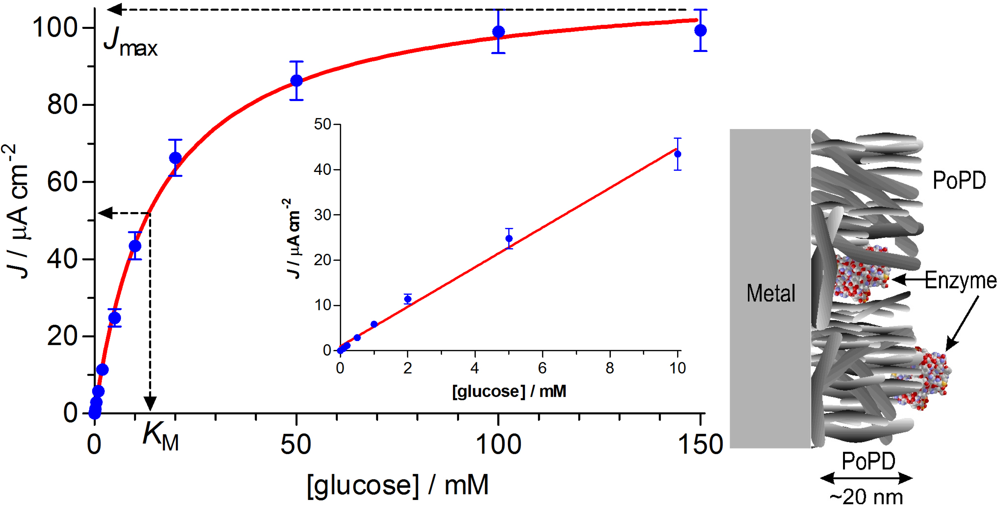
2.4. Enzyme Kinetic Parameters
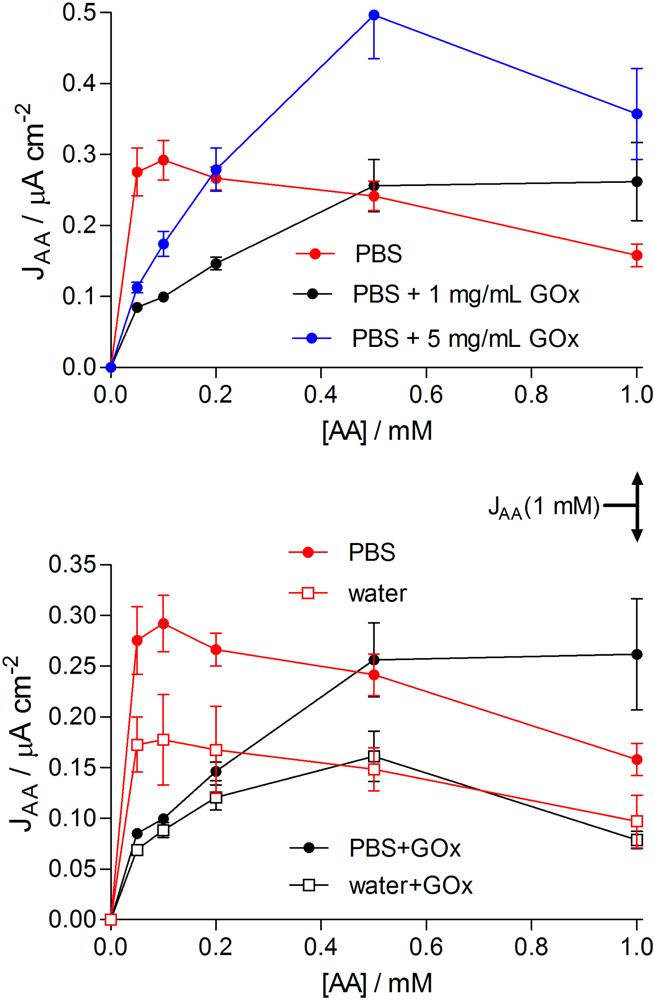
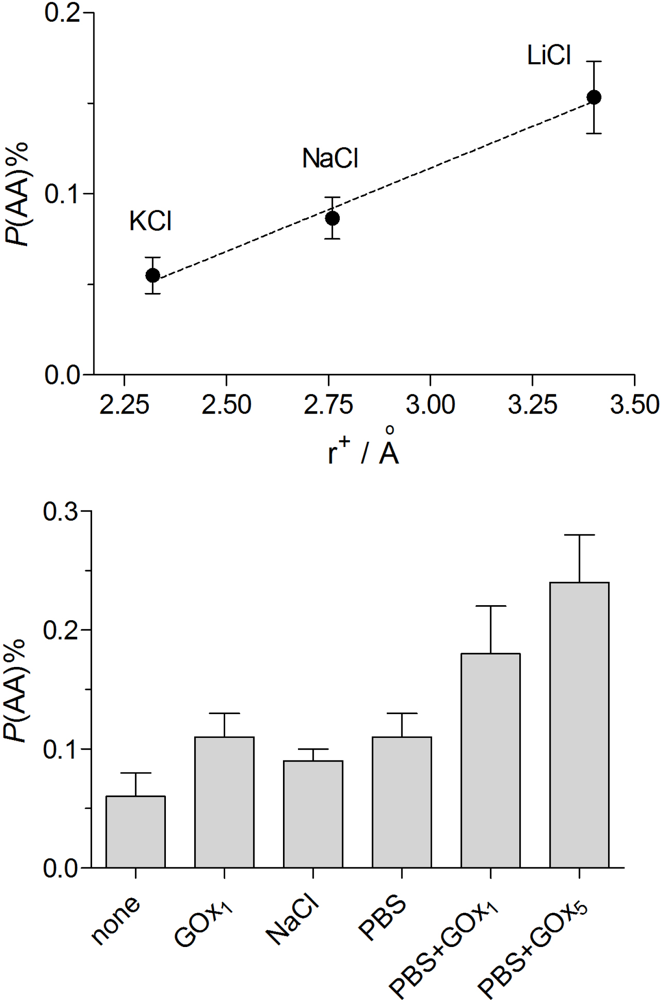
2.5. Permeability and Permselectivity Parameters
3. Results and Discussion
3.1. Michaelis-Menten Characteristics of the Basic Designs
3.2. Permeability Characteristics of the Basic Designs
3.3. Fine-Tuning the Conditions for Co-Immobilization of PoPD and GOx
3.3.1. Monomer Concentration
3.3.2. Enzyme Concentration
3.3.3. Electropolymerization Background Electrolyte
4. Conclusions
Acknowledgments
References and Notes
- Hirst, ER; Yuan, YJ; Xu, WL; Bronlund, JE. Bond-rupture immunosensors—A review. Biosens. Bioelectron 2008, 23, 1759–1768. [Google Scholar]
- Wanekaya, AK; Chen, W; Mulchandani, A. Recent biosensing developments in environmental security. J. Environ. Monit 2008, 10, 703–712. [Google Scholar]
- Sadik, OA; Aluoch, AO; Zhou, AL. Status of biomolecular recognition using electrochemical techniques. Biosens. Bioelectron 2009, 24, 2749–2765. [Google Scholar]
- Sozer, N; Kokini, JL. Nanotechnology and its applications in the food sector. Trends Biotechnol 2009, 27, 82–89. [Google Scholar]
- Saito, H; Nakazato, T; Ishii, N; Kudo, H; Otsuka, K; Endo, H; Mitsubayashi, K. An optical flow injection analysis system for measurement of glucose in tomato. Eur. Food Res. Technol 2008, 227, 473–478. [Google Scholar]
- Wei, D; Bailey, MJA; Andrew, P; Ryhanen, T. Electrochemical biosensors at the nanoscale. Lab Chip 2009, 9, 2123–2131. [Google Scholar]
- Mitsubayashi, K; Ohgoshi, T; Okamoto, T; Wakabayashi, Y; Kozuka, M; Miyajima, K; Saito, H; Kudo, H. Tonometric biosensor with a differential pressure sensor for chemo-mechanical measurement of glucose. Biosens. Bioelectron 2009, 24, 1518–1521. [Google Scholar]
- Lu, QZ; Lin, HL; Ge, ST; Luo, SL; Cai, QY; Grimes, CA. Wireless, remote-query, and high sensitivity escherichia coli O157:H7 biosensor based on the recognition action of Concanavalin A. Anal. Chem 2009, 81, 5846–5850. [Google Scholar]
- Tian, FM; Gourine, AV; Huckstepp, RTR; Dale, N. A microelectrode biosensor for real time monitoring of L-glutamate release. Anal. Chim. Acta 2009, 645, 86–91. [Google Scholar]
- Calia, G; Rocchitta, G; Migheli, R; Puggioni, GM; Spissu, Y; Bazzu, G; Mazzarello, V; Lowry, JP; O’Neill, RD; Desole, MS; Serra, PA. Biotelemetric monitoring of brain neurochemistry in conscious rats, using microsensors and biosensors. Sensors 2009, 9, 2511–2523. [Google Scholar]
- O’Neill, RD; Lowry, JP; Rocchitta, G; McMahon, CP; Serra, PA. Designing sensitive and selective polymer/enzyme composite biosensors for brain monitoring in vivo. Trends Anal. Chem 2008, 27, 78–88. [Google Scholar]
- Pernot, P; Mothet, JP; Schuvailo, O; Soldatkin, A; Pollegioni, L; Pilone, M; Adeline, MT; Cespuglio, R; Marinesco, S. Characterization of a yeast D-amino acid oxidase microbiosensor for D-serine detection in the central nervous system. Anal. Chem 2008, 80, 1589–1597. [Google Scholar]
- Wang, J. Electrochemical glucose biosensors. Chem. Rev 2008, 108, 814–825. [Google Scholar]
- Wilson, GS; Gifford, R. Biosensors for real-time in vivo measurements. Biosens. Bioelectron 2005, 20, 2388–2403. [Google Scholar]
- Burmeister, JJ; Gerhardt, GA. Ceramic-based multisite microelectrode arrays for in vivo electrochemical recordings of glutamate and other neurochemicals. Trends Anal. Chem 2003, 22, 498–502. [Google Scholar]
- Pantano, P; Kuhr, WG. Enzyme-modified microelectrodes for in vivo neurochemical measurements. Electroanalysis 1995, 7, 405–416. [Google Scholar]
- Lowry, JP; Ryan, MR; O’Neill, RD. Behaviourally induced changes in extracellular levels of brain glutamate monitored at 1 s resolution with an implanted biosensor. Anal. Commun 1998, 35, 87–89. [Google Scholar]
- Schuvailo, OM; Soldatkin, OO; Lefebvre, A; Cespuglio, R; Soldatkin, AP. Highly selective microbiosensors for in vivo measurement of glucose, lactate and glutamate. Anal. Chim. Acta 2006, 573, 110–116. [Google Scholar]
- Dai, YQ; Zhou, DM; Shiu, KK. Permeability and permselectivity of polyphenylenediamine films synthesized at a palladium disk electrode. Electrochim. Acta 2006, 52, 297–303. [Google Scholar]
- Carelli, I; Chiarotto, I; Curulli, A; Palleschi, G. Electropolymerization of hydroxybenzene and aminobenzene isomers on platinum electrodes to assemble interference-free electrochemical biosensors. Electrochim. Acta 1996, 41, 1793–1800. [Google Scholar]
- Li, XG; Huang, MR; Duan, W; Yang, YL. Novel multifunctional polymers from aromatic diamines by oxidative polymerisations. Chem. Rev 2002, 102, 2925–3030. [Google Scholar]
- Killoran, SJ; O’Neill, RD. Characterization of permselective coatings electrosynthesized on Pt-Ir from the three phenylenediamine isomers for biosensor applications. Electrochim. Acta 2008, 53, 7303–7312. [Google Scholar]
- McMahon, CP; Killoran, SJ; O’Neill, RD. Design variations of a polymer-enzyme composite biosensor for glucose: Enhanced analyte sensitivity without increased oxygen dependence. J. Electroanal. Chem 2005, 580, 193–202. [Google Scholar]
- Ryan, MR; Lowry, JP; O’Neill, RD. Biosensor for neurotransmitter L-glutamic acid designed for efficient use of L-glutamate oxidase and effective rejection of interference. Analyst 1997, 122, 1419–1424. [Google Scholar]
- Cooper, JM; Foreman, PL; Glidle, A; Ling, TW; Pritchard, DJ. Glutamate oxidase enzyme electrodes: microsensors for neurotransmitter determination using electrochemically polymerized permselective films. J. Electroanal. Chem 1995, 388, 143–149. [Google Scholar]
- Sasso, SV; Pierce, RJ; Walla, R; Yacynych, AM. Electropolymerized 1, 2-diaminobenzene as a means to prevent interferences and fouling and to stabilize immobilized enzyme in electrochemical biosensors. Anal. Chem 1990, 62, 1111–1117. [Google Scholar]
- McMahon, CP; Rocchitta, G; Serra, PA; Kirwan, SM; Lowry, JP; O’Neill, RD. Control of the oxygen dependence of an implantable polymer/enzyme composite biosensor for glutamate. Anal. Chem 2006, 78, 2352–2359. [Google Scholar]
- McMahon, CP; Rocchitta, G; Kirwan, SM; Killoran, SJ; Serra, PA; Lowry, JP; O’Neill, RD. Oxygen tolerance of an implantable polymer/enzyme composite glutamate biosensor displaying polycation-enhanced substrate sensitivity. Biosens. Bioelectron 2007, 22, 1466–1473. [Google Scholar]
- O’Brien, KB; Killoran, SJ; O’Neill, RD; Lowry, JP. Development and characterization in vitro of a catalase-based biosensor for hydrogen peroxide monitoring. Biosens. Bioelectron 2007, 22, 2994–3000. [Google Scholar]
- Lowry, JP; O’Neill, RD. Partial characterization in vitro of glucose oxidase-modified poly(phenylenediamine)-coated electrodes for neurochemical analysis in vivo. Electroanalysis 1994, 6, 369–379. [Google Scholar]
- Malitesta, C; Palmisano, F; Torsi, L; Zambonin, PG. Glucose fast-response amperometric sensor based on glucose oxidase immobilized in an electropolymerized poly(o-phenylenediamine) film. Anal. Chem 1990, 62, 2735–2740. [Google Scholar]
- Reyes De Corcuera, JI; Cavalieri, RP; Powers, JR. Improved platinization conditions produce a 60-fold increase in sensitivity of amperometric biosensors using glucose oxidase immobilized in poly-o-phenylenediamine. J. Electroanal. Chem 2005, 575, 229–241. [Google Scholar]
- Guilbault, GG. Analytical Uses of Immobilised Enzymes; Marcel Dekker: New York, NY, USA, 1984. [Google Scholar]
- Lee, CH; Wang, SC; Yuan, CJ; Wen, MF; Chang, KS. Comparison of amperometric biosensors fabricated by palladium sputtering, palladium electrodeposition and Nafion/carbon nanotube casting on screen-printed carbon electrodes. Biosens. Bioelectron 2007, 22, 877–884. [Google Scholar]
- Kulys, J; Drungiliene, A. Electrocatalytic oxidation of ascorbic acid at chemically modified electrodes. Electroanalysis 1991, 3, 209–214. [Google Scholar]
- El Atrash, SS; O’Neill, RD. Characterisation in vitro of a naphthoquinone-mediated glucose oxidase-modified carbon paste electrode designed for neurochemical analysis in vivo. Electrochim. Acta 1995, 40, 2791–2797. [Google Scholar]
- Lin, YQ; Liu, K; Yu, P; Xiang, L; Li, XC; Mao, LQ. A facille electrochemical method for simultaneous and on-line measurements of glucose and lactate in brain microdialysate with prussian blue as the electrocatalyst for reduction of hydrogen peroxide. Anal. Chem 2007, 79, 9577–9583. [Google Scholar]
- Fu, YC; Chen, C; Xie, QJ; Xu, XH; Zou, C; Zhou, QM; Tan, L; Tang, H; Zhang, YY; Yao, SZ. Immobilization of enzymes through one-pot chemical preoxidation and electropolymerization of dithiols in enzyme-containing aqueous suspensions to develop biosensors with improved performance. Anal. Chem 2008, 80, 5829–5838. [Google Scholar]
- Lowry, JP; McAteer, K; El Atrash, SS; Duff, A; O’Neill, RD. Characterization of glucose oxidase-modified poly(phenylenediamine)-coated electrodes in vitro and in vivo: Homogeneous interference by ascorbic acid in hydrogen peroxide detection. Anal. Chem 1994, 66, 1754–1761. [Google Scholar]
- Wang, J; Chen, L; Liu, J; Lu, F. Enhanced selectivity and sensitivity of first-generation enzyme electrodes based on the coupling of rhodinized carbon paste transducers and permselective poly(o-phenylenediamine) coatings. Electroanalysis 1996, 8, 1127–1130. [Google Scholar]
- Rothwell, SA; Kinsella, ME; Zain, ZM; Serra, PA; Rocchitta, G; Lowry, JP; O’Neill, RD. Contributions by a novel edge effect to the permselectivity of an electrosynthesized polymer for microbiosensor applications. Anal. Chem 2009, 81, 3911–3918. [Google Scholar]
- Rothwell, SA; Killoran, SJ; Neville, EM; Crotty, AM; O’Neill, RD. Poly(o-phenylenediamine) electrosynthesized in the absence of added background electrolyte provides a new permselectivity benchmark for biosensor applications. Electrochem. Commun 2008, 10, 1078–1081. [Google Scholar]
- Guerrieri, A; Lattanzio, V; Palmisano, F; Zambonin, PG. Electrosynthesized poly(pyrrole)/poly(2-naphthol) bilayer membrane as an effective anti-interference layer for simultaneous determination of acethylcholine and choline by a dual electrode amperometric biosensor. Biosens. Bioelectron 2006, 21, 1710–1718. [Google Scholar]
- Kirwan, SM; Rocchitta, G; McMahon, CP; Craig, JD; Killoran, SJ; O’Brien, KB; Serra, PA; Lowry, JP; O’Neill, RD. Modifications of poly(o-phenylenediamine) permselective layer on Pt-Ir for biosensor application in neurochemical monitoring. Sensors 2007, 7, 420–437. [Google Scholar]
- Craig, JD; O’Neill, RD. Comparison of simple aromatic amines for electrosynthesis of permselective polymers in biosensor fabrication. Analyst 2003, 128, 905–911. [Google Scholar]
- Myler, S; Eaton, S; Higson, SPJ. Poly(o-phenylenediamine) ultra-thin polymer-film composite membranes for enzyme electrodes. Anal. Chim. Acta 1997, 357, 55–61. [Google Scholar]
- Gooding, JJ; Hall, EAH. Parameters in the design of oxygen detecting oxidase enzyme electrodes. Electroanalysis 1996, 8, 407–413. [Google Scholar]
- Dixon, BM; Lowry, JP; O’Neill, RD. Characterization in vitro and in vivo of the oxygen dependence of an enzyme/polymer biosensor for monitoring brain glucose. J. Neurosci. Meth 2002, 119, 135–142. [Google Scholar]
- Compagnone, D; Federici, G; Bannister, JV. A new conducting polymer glucose sensor based on polythianaphthene. Electroanalysis 1996, 7, 1151–1155. [Google Scholar]
- Centonze, D; Guerrieri, A; Malitesta, C; Palmisano, F; Zambonin, PG. An in-situ electrosynthesized poly-ortho-phenylenediamine/glucose oxidase amperometric biosensor for flow-injection determination of glucose in serum. Ann. Chim. (Rome) 1992, 82, 219–234. [Google Scholar]
- Miele, M; Fillenz, M. In vivo determination of extracellular brain ascorbate. J. Neurosci. Meth 1996, 70, 15–19. [Google Scholar]
- Boutelle, MG; Svensson, L; Fillenz, M. Rapid changes in striatal ascorbate in response to tail-pinch monitored by constant potential voltammetry. Neuroscience 1989, 30, 11–17. [Google Scholar]
- O’Neill, RD; Fillenz, M; Albery, WJ. The development of linear sweep voltammetry with carbon paste electrodes in vivo. J. Neurosci. Meth 1983, 8, 263–273. [Google Scholar]
- Fillenz, M; O’Neill, RD. Effects of light reversal on the circadian pattern of motor activity and voltammetric signals recorded in rat forebrain. J. Physiol. (London) 1986, 374, 91–101. [Google Scholar]
- Brose, N; O’Neill, RD; Boutelle, MG; Anderson, SMP; Fillenz, M. Effects of an anxiogenic benzodiazepine receptor ligand on rat motor activity and dopamine release in nucleus accumbens and striatum. J. Neurosci 1987, 7, 2917–2926. [Google Scholar]
- Soldatkin, OO; Schuvailo, OM; Marinesco, S; Cespuglio, R; Soldatkin, AR. Microbiosensor based on glucose oxidase and hexokinase co-immobilised on platinum microelectrode for selective ATP detection. Talanta 2009, 78, 1023–1028. [Google Scholar]
- Santos, RM; Lourenco, CF; Piedade, AP; Andrews, R; Pomerleau, F; Huettl, P; Gerhardt, GA; Laranjinha, J; Barbosa, RM. A comparative study of carbon fiber-based microelectrodes for the measurement of nitric oxide in brain tissue. Biosens. Bioelectron 2008, 24, 704–709. [Google Scholar]
- Hamdi, N; Wang, JJ; Monbouquette, HG. Polymer films as permselective coatings for H2O2-sensing electrodes. J. Electroanal. Chem 2005, 581, 258–264. [Google Scholar]
- McAteer, K; O’Neill, RD. Strategies for decreasing ascorbate interference at glucose oxidase-modified poly(o-phenylenediamine)-coated electrodes. Analyst 1996, 121, 773–777. [Google Scholar]
- Lowry, JP; O’Neill, RD; Boutelle, MG; Fillenz, M. Continuous monitoring of extracellular glucose concentrations in the striatum of freely moving rats with an implanted glucose biosensor. J. Neurochem 1998, 70, 391–396. [Google Scholar]
- Dai, YQ; Shiu, KK. Highly sensitive amperometric glucose biosensor based on glassy carbon electrode with copper/palladium coating. Electroanalysis 2004, 16, 1806–1813. [Google Scholar]
- Bartlett, PN; Wang, JH; James, W. Measurement of low glucose concentrations using a microelectrochemical enzyme transistor. Analyst 1998, 123, 387–392. [Google Scholar]
- Wang, J; Lu, F. Oxygen-rich oxidase enzyme electrodes for operation in oxygen-free solutions. J. Am. Chem. Soc 1998, 120, 1048–1050. [Google Scholar]
- Wang, J. Selectivity coefficients for amperometric sensors. Talanta 1994, 41, 857–863. [Google Scholar]
- Palmisano, F; Rizzi, R; Centonze, D; Zambonin, PG. Simultaneous monitoring of glucose and lactate by an interference and cross-talk free dual electrode amperometric biosensor based on electropolymerized thin films. Biosens. Bioelectron 2000, 15, 531–539. [Google Scholar]
- Ahmad, F; Christenson, A; Bainbridge, M; Yusof, APM; Ab Ghani, S. Minimizing tissue-material interaction in microsensor for subcutaneous glucose monitoring. Biosens. Bioelectron 2007, 22, 1625–1632. [Google Scholar]
- Maalouf, R; Chebib, H; Saikali, Y; Vittori, O; Sigaud, M; Garrelie, F; Donnet, C; Jaffirezic-Renault, N. Characterization of different diamond-like carbon electrodes for biosensor design. Talanta 2007, 72, 310–314. [Google Scholar]
- Cooper, JM; Pritchard, DJ. Biomolecular sensors for neurotransmitter determination electrochemical immobilization of glutamate oxidase at microelectrodes in a poly (o-phenylenediamine) film. J. Mater. Sci.: Mater. Electronl 1994, 5, 111–116. [Google Scholar]
- Bao, L; Avshalumov, MV; Patel, JC; Lee, CR; Miller, EW; Chang, CJ; Rice, ME. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci 2009, 29, 9002–9010. [Google Scholar]
- McMahon, CP; Rocchitta, G; Serra, PA; Kirwan, SM; Lowry, JP; O’Neill, RD. The efficiency of immobilised glutamate oxidase decreases with surface enzyme loading: an electrostatic effect, and reversal by a polycation significantly enhances biosensor sensitivity. Analyst 2006, 131, 68–72. [Google Scholar]
- Burmeister, JJ; Palmer, M; Gerhardt, GA. Ceramic-based multisite microelectrode array for rapid choline measures in brain tissue. Anal. Chim. Acta 2003, 481, 65–74. [Google Scholar]
- Berners, MOM; Boutelle, MG; Fillenz, M. On-line measurement of brain glutamate with an enzyme/polymer-coated tubular electrode. Anal. Chem 1994, 66, 2017–2021. [Google Scholar]
- Kulagina, NV; Shankar, L; Michael, AC. Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal. Chem 1999, 71, 5093–5100. [Google Scholar]
- Harrar, JE. Controlled-potential coulometric determination of hydrogen peroxide. Anal. Chem 1963, 35, 893–896. [Google Scholar]
- Rothwell, SA; McMahon, CP; O’Neill, RD. Effects of polymerization potential on the permselectivity of poly(o-phenylenediamine) coatings deposited on Pt-Ir electrodes for biosensor applications. Electrochim. Acta 2010, 55, 1051–1060. [Google Scholar]
- Hascup, KN; Hascup, ER; Pomerleau, F; Huettl, P; Gerhardt, GA. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J. Pharmacol. Exp. Ther 2008, 324, 725–731. [Google Scholar]
- Morales-Villagran, A; Medina-Ceja, L; Lopez-Perez, SJ. Simultaneous glutamate and EEG activity measurements during seizures in rat hippocampal region with the use of an electrochemical biosensor. J. Neurosci. Meth 2008, 168, 48–53. [Google Scholar]
- Du, D; Ding, JW; Cai, J; Zhang, AD. One-step electrochemically deposited interface of chitosan-gold nanoparticles for acetylcholinesterase biosensor design. J. Electroanal. Chem 2007, 605, 53–60. [Google Scholar]
- Dale, N; Hatz, S; Tian, FM; Llaudet, E. Listening to the brain: microelectrode biosensors for neurochemicals. Trends Biotechnol 2005, 23, 420–428. [Google Scholar]
- Patel, BA; Arundell, M; Parker, KH; Yeoman, MS; O’Hare, D. Detection of nitric oxide release from single neurons in the pond snail, Lymnaea stagnalis. Anal. Chem 2006, 78, 7643–7648. [Google Scholar]
- Losito, I; Palmisano, F; Zambonin, PG. o-Phenylenediamine electropolymerization by cyclic voltammetry combined with electrospray ionization-ion trap mass spectrometry. Anal. Chem 2003, 75, 4988–4995. [Google Scholar]
- Palmisano, F; Zambonin, PG; Centonze, D. Amperometric biosensors based on electrosynthesised polymeric films. Fresenius J. Anal. Chem 2000, 366, 586–601. [Google Scholar]
- Centonze, D; Losito, I; Malitesta, C; Palmisano, F; Zambonin, PG. Electrochemical immobilisation of enzymes on conducting organic salt electrodes: characterisation of an oxygen independent and interference-free glucose biosensor. J. Electroanal. Chem 1997, 435, 103–111. [Google Scholar]
- Wang, Q; Tang, H; Xie, QJ; Jia, XE; Zhang, YY; Tan, L; Yao, SZ. The preparation and characterization of poly(o-phenylenediamine)/gold nanoparticles interface for immunoassay by surface plasmon resonance and electrochemistry. Colloid. Surface. B 2008, 63, 254–261. [Google Scholar]
- Camurri, G; Ferrarini, P; Giovanardi, R; Benassi, R; Fontanesi, C. Modelling of the initial stages of the electropolymerization mechanism of o-phenylenediamine. J. Electroanal. Chem 2005, 585, 181–190. [Google Scholar]
- Losito, I; De Giglio, E; Cioffi, N; Malitesta, C. Spectroscopic investigation on polymer films obtained by oxidation of o-phenylenediamine on platinum electrodes at different pHs. J. Mater. Chem 2001, 11, 1812–1817. [Google Scholar]
- Ekinci, E; Erdogdu, G; Karagozler, AE. Preparation, optimization, and voltammetric characteristics of poly(o-phenylenediamine) film as a dopamine-selective polymeric membrane. J. Appl. Polym. Sci 2001, 79, 327–332. [Google Scholar]
- Centonze, D; Malitesta, C; Palmisano, F; Zambonin, PG. Permeation of solutes through an electropolymerized ultrathin poly-o-phenylenediamine film used as an enzyme-entrapping membrane. Electroanalysis 1994, 6, 423–429. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Design | n-value | Jmax (μA cm−2) | KM (mM) | LRS (μA cm−2 mM−1) | BE% (%) |
|---|---|---|---|---|---|
| PtC/GOx-GA | 4 | 66 ± 5 | 13 ± 1 | 3.6 ± 0.2 | 1.4 ± 0.1 |
| PtC/GOx-GA/PoPD | 4 | 33 ± 3 | 32 ± 3 | 1.2 ± 0.1 | 0.47 ± 0.05 |
| PtC/GOx/PoPD | 7 | 10 ± 1 | 21 ± 2 | 0.37 ± 0.03 | 0.17 ± 0.01 |
| PtC/PoPD/GOx-GA | 4 | 72 ± 3 | 26 ± 2 | 2.7 ± 0.1 | 0.66 ± 0.02 |
| PtC/GOx/PoPD/GOx-GA | 4 | 50 ± 5 | 26 ± 3 | 1.7 ± 0.4 | 0.55 ± 0.07 |
| PtC/PoPD-GOx | 8 | 111 ± 6 | 16 ± 2 | 5.0 ± 0.4 | 2.0 ± 0.2 |
| Design | n-value | P(HP)% | P(AA)% | S% | SG% |
|---|---|---|---|---|---|
| PtC/PoPD | 19 | 90 ± 5 | 0.11 ± 0.02 | 0.09 ± 0.01 | N/A* |
| PtC/GOx/PoPD | 6 | 97 ± 7 | 0.34 ± 0.05 | 0.20 ± 0.03 | 139 ± 36 |
| PtC/PoPD/GOx-GA | 4 | 98 ± 2 | 0.51 ± 0.07 | 0.29 ± 0.03 | 29 ± 8 |
| PtC/GOx/PoPD/GOx-GA | 4 | 127 ± 8 | 1.0 ± 0.1 | 0.55 ± 0.07 | 108 ± 32 |
| PtC/PPD-GOx | 8 | 120 ± 7 | 0.24 ± 0.04 | 0.14 ± 0.02 | 7 ± 1 |
| Background electrolyte: | PBS | PBS | KCl | none |
|---|---|---|---|---|
| [GOx] (mg mL−1): | 5.0 | 1.0 | 1.0 | 1.0 |
| Jmax (μA cm−2) | 111 ± 6 | 85 ± 7 | 87 ± 3 | 96 ± 5 |
| KM (mM) | 16 ± 2 | 13 ± 2 | 27 ± 1 | 10 ± 1 |
| LRS (μA cm−2 mM−1) | 5.0 ± 0.4 | 5.0 ± 1.0 | 3.1 ± 0.2 | 6.5 ± 0.6 |
| BE% | 2.0 ± 0.2 | 2.0 ± 0.4 | 1.1 ± 0.1 | 2.4 ± 0.2 |
| P(AA)% | 0.24 ± 0.04 | 0.18 ± 0.04 | 0.11 ± 0.01 | 0.11 ± 0.02 |
| S% | 0.14 ± 0.02 | 0.09 ± 0.02 | 0.06 ± 0.01 | 0.07 ± 0.01 |
| SG% | 7 ± 1 | 7 ± 2 | 6 ± 1 | 2 ± 1 |
© 2010 by the authors licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rothwell, S.A.; Killoran, S.J.; O’Neill, R.D. Enzyme Immobilization Strategies and Electropolymerization Conditions to Control Sensitivity and Selectivity Parameters of a Polymer-Enzyme Composite Glucose Biosensor. Sensors 2010, 10, 6439-6462. https://doi.org/10.3390/s100706439
Rothwell SA, Killoran SJ, O’Neill RD. Enzyme Immobilization Strategies and Electropolymerization Conditions to Control Sensitivity and Selectivity Parameters of a Polymer-Enzyme Composite Glucose Biosensor. Sensors. 2010; 10(7):6439-6462. https://doi.org/10.3390/s100706439
Chicago/Turabian StyleRothwell, Sharon A., Sarah J. Killoran, and Robert D. O’Neill. 2010. "Enzyme Immobilization Strategies and Electropolymerization Conditions to Control Sensitivity and Selectivity Parameters of a Polymer-Enzyme Composite Glucose Biosensor" Sensors 10, no. 7: 6439-6462. https://doi.org/10.3390/s100706439