Voltammetry under a Controlled Temperature Gradient

Abstract
:
1. Introduction
Theory
| σ | is the local entropy production, |
| Jq | is the flow of heat, |
| T | is the absolute temperature, |
| Ji | is the flow of component i in moles per unit area per unit time, and |
| μi | is the chemical potential of component i. |
is the flow of the solute relative to the solvent in binary solutions, | |
| Lik | is a phenomenological coefficient relating to the kth driving force (L11, L1q where q is heat), |
| μss | is the chemical potential of the solute in a binary system, and |
| Cs | is the molar concentration of the solute in a binary solution. |
Theory of the potential steps at an planar electrode including thermo diffusion mass transport
| CO(x,t) | is the concentration of the oxidized compound (reducible), |
| CR(x,t) | is the concentration of the reduced compound (oxidable), |
| n | is the number of electrons in the electrode reaction, |
| F | is the Faraday constant, |
| E | is the electrode potential, |
| E0 | is the standard electrode reaction potential, |
| R | is the gas constant, |
| T1 | is the temperature of the electrode surface, and |
| t | is the time. |
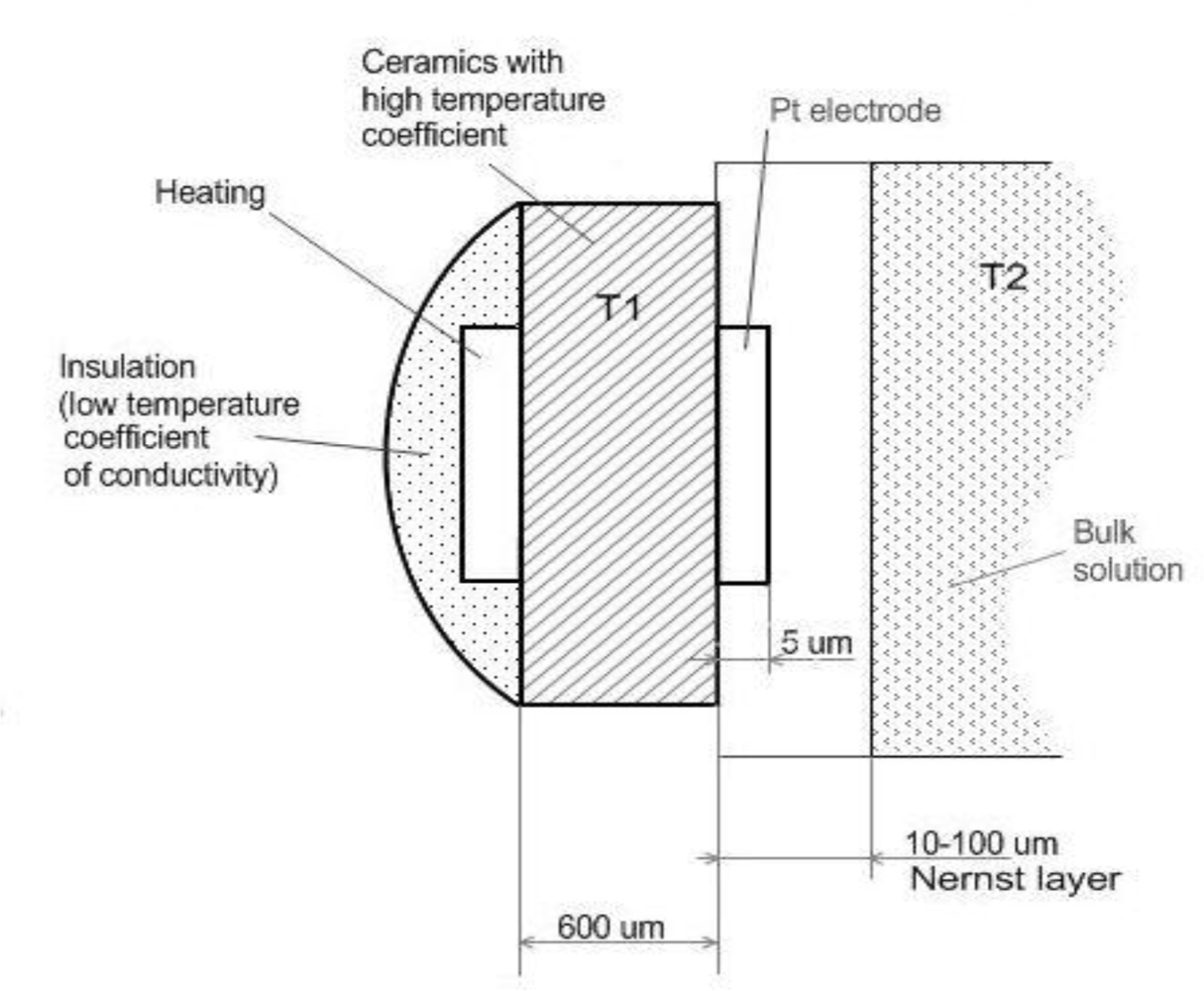
- - The electrode is planar and has a surface area A. Only diffusion along the x-axis perpendicular to the electrode surface needs to be considered. The electrode is sufficiently large so that the edge effects can be neglected.
- - The solution is initially homogenous. Specifically, the initial concentration of O and R are CO(x,0) = CO and CR(x,0) = 0 for all values of x at time t = 0.
- - The electrolysis cell is sufficiently large that the bulk concentrations of O and R are unchanged from the initial values even after electrolysis has been running for a certain time. In other words, CO(x,t)→CO, CR(x,t)→0 while x→∞.
- - For every O molecule consumed, an R molecule is formed; in other words, the fluxes JO(0,t) and JR(0,t) of O and R at the electrode surface are equal and opposite in sign: JO(0,t) = −JR(0,t).
- - The electron-transfer reaction is very fast, such that O and R are always in equilibrium at the electrode surface with the concentration ratio given by the Nernst Equation (7).
- - The system can be treated as two binary systems of (solvent and O) and (solvent and R). There is no interaction between O and R.
- - The reaction takes place at the electrode surface, the temperature of which is T1.
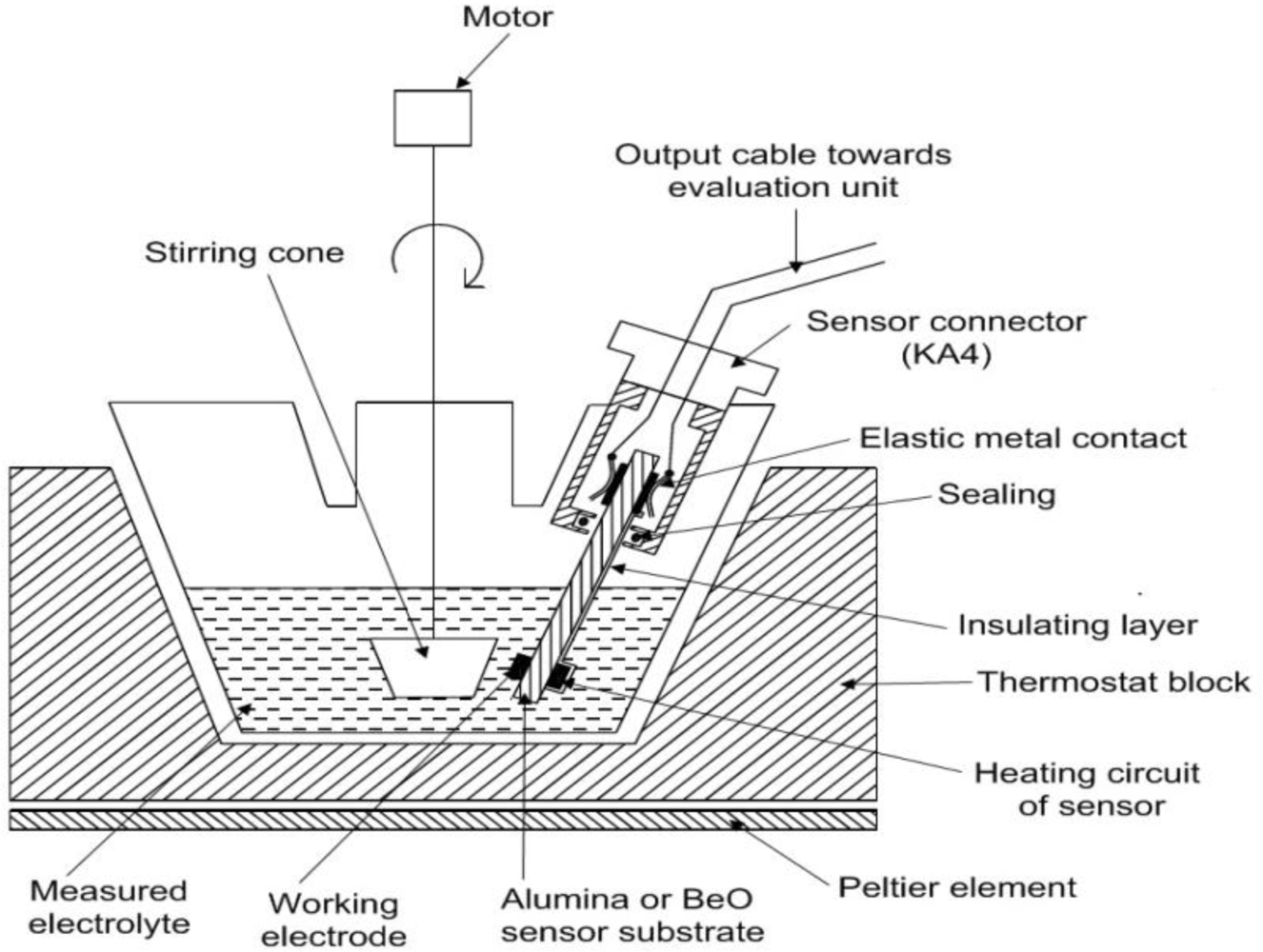
2. Experimental Section
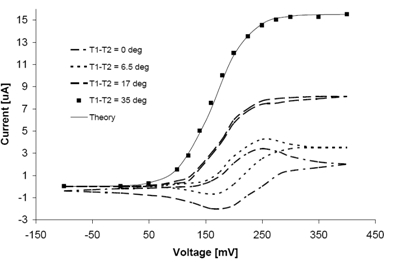
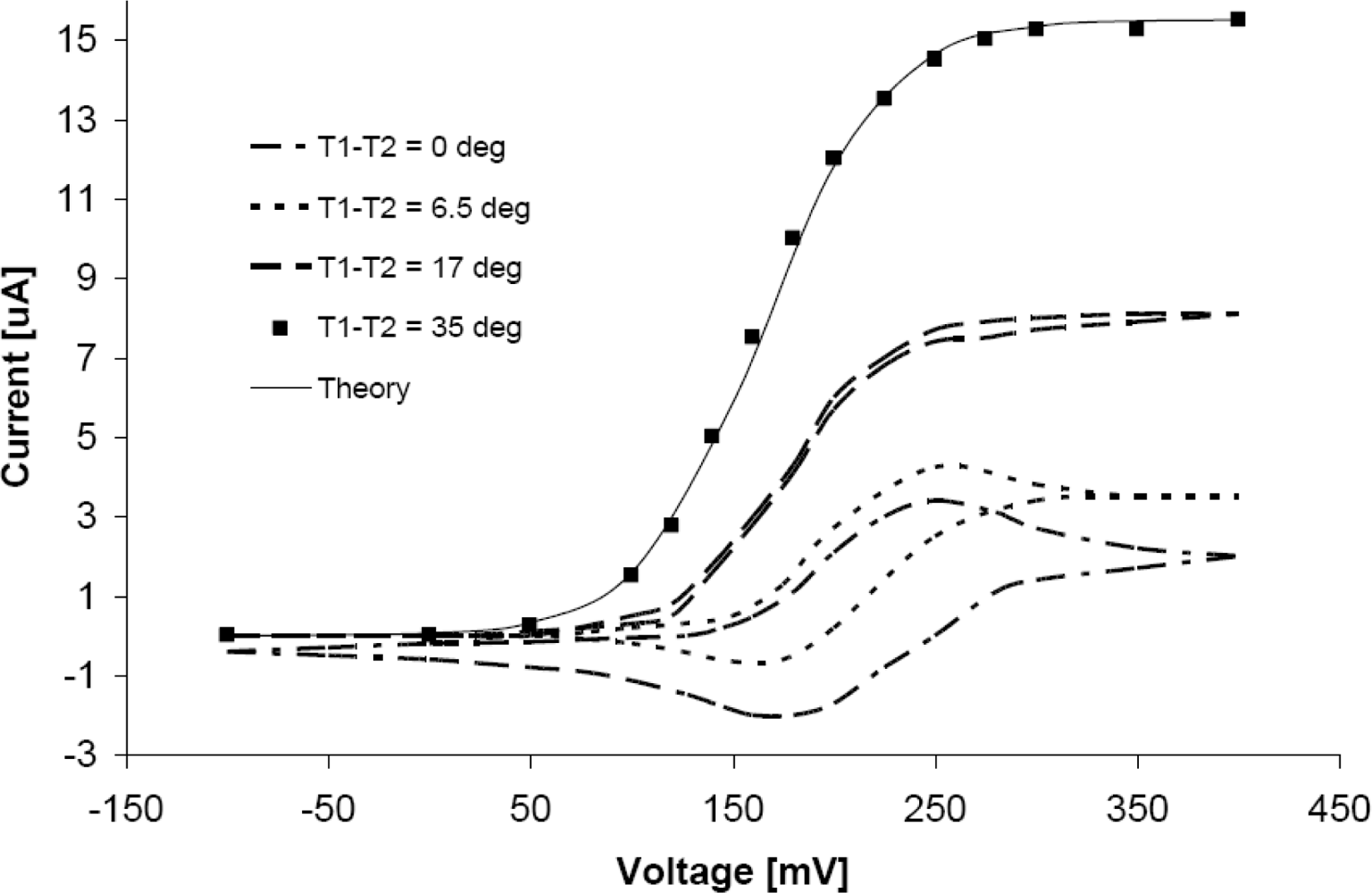
3. Results and Discussion
4. Conclusions
List of Symbols
| A | area of electrode surface |
| CO(x,t) | the concentration of oxidized compound (reducible) |
| CR(x,t) | the concentration of reduced compound (oxidable) |
| Cs | molar concentration of solute in a binary solution |
| C0 | initial molar concentration |
| DOx, DRed | diffusion coefficients for Ox and Red, respectively |
| E | cell potential |
| E0 | standard redox potential |
| F | Faraday constant |
| i | electric current |
| i0 | steady state current |
| J | molar flux density |
| Ji | flow of component i in moles per unit area per unit time |
| Jq | is a flow of heat |
| Jsd | flow of solute relative to solvent in binary solutions |
| Lik | phenomenological coefficient relating to the kth driving force (L11, L1q where q is heat) |
| λ | specific heat conductivity |
| n | number of electrons in electrode reaction |
| R | gas constant |
| sT, Ox and sT,Red | Soret coefficients for Ox and Red, respectively |
| t | time |
| T | absolute temperature |
| T1 | temperature of electrode surface |
| T2 | temperature of liquid |
| α | proportionality constant |
| μi | chemical potential of component i |
| μss | chemical potential of solute in a binary system |
Acknowledgments
References
- Bard, AJ; Faulkner, LR. Electrochemical Methods–Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 1980. [Google Scholar]
- Rieger, PH. Electrochemistry; Prentice Hall: Engelwood Cliffs, NJ, USA, 1987. [Google Scholar]
- Leal, LG. Advanced Transport Phenomena: Fluid Mechanics and Convective Transport Processes; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Bird, RB; Steward, WE; Lightfoot, EN. Transfer Phenomena, 2nd ed; John Wiley & Sons: Toronto, Canada, 2001; p. 912. [Google Scholar]
- Frost, W; Moulden, TH (Eds.) Handbook of Turbulence Volume 1 Fundamentals and Applications; Plenum Press: New York, NY, USA, 1977; p. 498.
- Gründler, P; Zerihun, T; Kirbs, A; Grabow, H. Simultaneous joule heating and potential cycling of cylindrical microelectrodes. Anal. Chim. Acta 1995, 305, 232–240. [Google Scholar]
- Smalley, JF; Geng, L; Feldberg, SW; Rogers, LC; Leddy, J. Evidence for adsorption of Fe(CN)63−/4− on gold using the indirect laser-induced temperature-jump method. J. Electroanal. Chem 1993, 356, 181–200. [Google Scholar]
- Harima, Y; Aoyagui, S. Electrode potential relaxation following a rapid change of temperature: Part II. Theory. J. Electroanal. Chem 1977, 81, 47–52. [Google Scholar]
- Harima, Y; Aoyagui, S. Electrode potential relaxation following a rapid change of temperature. J. Electroanal. Chem 1976, 69, 419–422. [Google Scholar]
- Gründler, P; Zerihun, T. Electrically heated cylindrical microelectrodes. The reduction of dissolved oxygen on Pt. J. Electroanal. Chem 1996, 404, 234–238. [Google Scholar]
- Frischmuth, K; Visocky, P; Gründler, P. On modeling heat transfer in chemical microsensors. Int. J. Eng. Sci 1996, 34, 523–530. [Google Scholar]
- Green, NG; Ramos, A; González, A; Castellanos, A; Morgan, H. Electrothermally induced fluid flow on microelectrodes. J. Electrostat 2001, 53, 1–87. [Google Scholar]
- Oduoza, CF. Electrochemical mass transfer at a heated electrode in a vertical annular flow cell. Chem. Eng. Process 2004, 43, 921–928. [Google Scholar]
- Sun, JJ; Guo, L; Zhang, DF; Yin, WH; Chen, GN. Heated graphite cylinder electrodes. Electrochem. Commun 2007, 9, 283–288. [Google Scholar]
- Mahnke, N; Markovic, A; Duwensee, H; Wachholz, F; Flechsig, GU; van Rienen, U. Numerically optimized shape of directly heated electrodes for minimal temperature gradients. Sens. Actuat. B 2009, 137, 363–369. [Google Scholar]
- Zerihun, T; Gründler, P. Electrically heated cylindrical microelectrodes. Determination of lead on Pt by cyclic voltammetry and cathodic stripping analysis. J. Electroanal. Chem 1996, 415, 85–88. [Google Scholar]
- Zerihun, T; Gründler, P. Oxidation of formaldehyde, methanol, formic acid and glucose at ac heated cylindrical Pt microelectrodes. J. Electroanal. Chem 1998, 441, 57–63. [Google Scholar]
- Jasinski, M; Kirbs, A; Schmehl, M; Gründler, P. Heated mercury film electrode for anodic stripping voltammetry. Electrochem. Commun 1999, 1, 26–28. [Google Scholar]
- Voss, T; Gründler, P; Brett, CMA; Oliveira Brett, AM. Electrochemical behavior of cytochrome c at electrically heated microelectrodes. J. Pharm. Biomed. Anal 1999, 19, 127–133. [Google Scholar]
- Korbut, O; Buckova, M; Tarapcik, P; Labuda, J; Gründler, P. Damage to DNA indicated by an electrically heated DNA—modified carbon paste electrode. J. Electroanal. Chem 2001, 506, 143–148. [Google Scholar]
- Wang, J; Gründler, P. The influence of temperature on the interaction between DNA and metal complec at a heated gold-wire microelectrode. J. Electroanal. Chem 2003, 540, 153–157. [Google Scholar]
- Lau, C; Flechsig, GU; Gründler, P; Wang, J. Electrochemistry of nicotinamide adenine dinucleotide (reduced) at heated platinum electrodes. Anal. Chim. Acta 2005, 554, 74–78. [Google Scholar]
- Lau, C; Borgmann, S; Maciejewska, M; Ngounou, B; Gründler, P; Schuhmann, W. Improved specificity of reagentless amperometric PQQ-sGDH glucose biosensors by using indirectly heated electrodes. Biosens. Bioelectron 2007, 22, 3014–3020. [Google Scholar]
- Ke, JH; Tseng, HJ; Hsu, ChT; Chen, JCh; Muthuraman, G; Zen, JM. Flow injection analysis of ascorbic acid based on its thermoelectrochemistry at disposable screen-printed carbon electrodes. Sens. Actuators, B 2008, 130, 614–619. [Google Scholar]
- Wu, SH; Sun, JJ; Zhang, DF; Lin, ZB; Nie, FH; Qiu, HY; Chen, GN. Nanomolar detection of rutin based on adsorptive stripping analysis at single-sided heated graphite cylindrical electrodes with direct current heating. Electrochim. Acta 2008, 53, 6596–6601. [Google Scholar]
- Tseng, TF; Yang, YL; Chuang, MC; Lou, SL; Galik, M; Flechsig, GU; Wang, J. Thermally stable improved first-generation glucose biosensors based on Nafion/glucose-oxidase modified heated electrodes. Electrochem. Commun 2009, 11, 1819–1822. [Google Scholar]
- Duwensee, H; Vázquez-Alvarez, T; Flechsig, GU; Wang, J. Thermally induced electrode protection against biofouling. Talanta 2009, 77, 1757–1760. [Google Scholar]
- Chen, Y; Lin, Z; Chen, J; Sun, J; Zhang, L; Chen, G. New capillary electrophoresis-electrochemiluminescence detection system equipped with an electrically heated Ru(bpy)32+/multi-wall-carbon-nanotube paste electrode. J. Chromatogr. A 2007, 1172, 84–91. [Google Scholar]
- Wang, J; Jasinski, M; Flechsig, GU; Grundler, P; Tian, B. Hot-wire amperometric monitoring of flowing streams. Talanta 2000, 50, 1205–1210. [Google Scholar]
- Jenkins, DM; Song, C; Fares, S; Cheng, H; Barrettino, D. Disposable thermostated electrode system for temperature dependent electrochemical measurements. Sens. Actuator. B 2009, 137, 222–229. [Google Scholar]
- Voß, T; Gründler, P; Kirbs, A; Flechsig, GU. Temperature pulse voltammetry: hot layer electrodes made by LTCC technology. Electrochem. Commun 1999, 1, 383–388. [Google Scholar]
- Wang, J; Gründler, P; Flechsig, GU; Jasinski, M; Lu, J; Wang, J; Zhao, Z; Tian, B. Hot-wire stripping potentiometric measurements of trace mercury. Anal. Chim. Acta 1999, 396, 33–37. [Google Scholar]
- Qiu, F; Compton, RG; Coles, BA; Marken, F. Thermal activation of electrochemical processes in a Rf-heated channel flow cell: experiment and finite element simulation. J. Electroanal. Chem 2000, 492, 150–155. [Google Scholar]
- Coles, BA; Moorcroft, MJ; Compton, RG. Non – aqueous electrochemical studies at a high temperature channel flow cell heated by radio frequency radiation. J. Electroanal. Chem 2001, 513, 87–93. [Google Scholar]
- Wachholz, F; Biała, K; Piekarz, M; Flechsig, GU. Temperature pulse modulated amperometry at compact electrochemical sensors. Electrochem. Commun 2007, 9, 2346–2352. [Google Scholar]
- Xu, H; Xing, S; Zeng, L; Xian, Y; Shi, G; Jin, L. Microwave-enhanced voltammetric detection of copper(II) at gold nanoparticles-modified platinum microelectrodes. J. Electroanal. Chem 2009, 625, 53–59. [Google Scholar]
- Lin, Z; Sun, J; Chen, J; Guo, L; Chen, G. A new electrochemiluminescent detection system equipped with an electrically controlled heating cylindrical microelectrode. Anal. Chim. Acta 2006, 564, 226–230. [Google Scholar]
- Lin, Z; Sun, J; Chen, J; Guo, Li; Chen, G. The electrochemiluminescent behavior of luminol on an electrically heating controlled microelectrode at cathodic potential. Electrochim. Acta 2007, 53, 1708–1712. [Google Scholar]
- Chen, Y; Chen, X; Lin, Z; Dai, H; Qiu, B; Sun, J; Zhang, L; Chen, G. An electrically heated ionic-liquid/multi-wall carbon nanotube composite electrode and its application to electrochemiluminescent detection of ascorbic acid. Electrochem. Commun 2009, 11, 1142–1145. [Google Scholar]
- Lin, Z; Chen, Xi; Chen, H; Qiu, B; Chen, G. Electrochemiluminescent behavior of N6-isopentenyl-adenine/Ru(bpy)32+ system on an electrically heated ionic liquid/carbon paste electrode. Electrochem. Commun 2009, 11, 2056–2059. [Google Scholar]
- Li, Y; Liu, X; Zeng, X; Liu, Y; Liu, Xi; Wei, W; Luo, S. Simultaneous determination of ultra-trace lead and cadmium at a hydroxyapatite-modified carbon ionic liquid electrode by square-wave stripping voltammetry. Sens. Actuat. B 2009, 139, 604–610. [Google Scholar]
- Katchalsky, A; Curran, PF. Nonequilibrium Thermodynamics in Biophysics; Harvard University Press: Cambridge, MA, USA, 1967. [Google Scholar]
- Jost, J. Diffusion in Solids, Liquid and Gases; Academic Press: New York, NY, USA, 1960. [Google Scholar]
- Bringuier, E; Bourdon, A. Kinetic theory of colloid thermodiffusion. Physica A 2007, 385, 9–24. [Google Scholar]
- Bringuier, E. Kinetic theory of inhomogeneous diffusion. Physica A 2009, 388, 2588–2599. [Google Scholar]
- Lorenz, EN. Deterministic Nonperiodic Flow. J. Atmos. Sci 1963, 20, 130–141. [Google Scholar]
- Sparrow, C. Applied mathematical sciences. In The Lorenz Equations: Bifurcations, Chaos and Strange Attractors; Springer-Verlag: New York, NY, USA, 1982. [Google Scholar]
- Holodniok, M; Klic, A; Kubicek, M; Marek, M. Metody analyzy nelinearnich dynamickych modelu; Academia: Prague, Czechoslovakia, 1986. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | λ | α | D |
|---|---|---|---|
| W/m·K | m2/s | m2/s | |
| Water at 25 °C | 0.6 | 0.14 × 10−6 | 10−10–10−12* |
| Al2O3 ceramic | 35 | 8.8 × 10−6 | |
| BeO | 180 | 89 × 10−6 | |
| Ag | 420 | 172 × 10−6 |
© 2010 by the authors licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krejci, J.; Sajdlova, Z.; Krejci, J., Jr.; Marvanek, T. Voltammetry under a Controlled Temperature Gradient. Sensors 2010, 10, 6821-6835. https://doi.org/10.3390/s100706821
Krejci J, Sajdlova Z, Krejci J Jr., Marvanek T. Voltammetry under a Controlled Temperature Gradient. Sensors. 2010; 10(7):6821-6835. https://doi.org/10.3390/s100706821
Chicago/Turabian StyleKrejci, Jan, Zuzana Sajdlova, Jan Krejci, Jr., and Tomas Marvanek. 2010. "Voltammetry under a Controlled Temperature Gradient" Sensors 10, no. 7: 6821-6835. https://doi.org/10.3390/s100706821